Primárne imunodeficiencie definície znakov klasifikácie PID. Imunodeficitné choroby
Imunodeficiencia(ID) je genetický a/alebo laboratórny príznak defektu (nedostatočnosti) väzby imunity s klinickým stavom alebo bez neho klinické prejavy.
Bežné príznaky ochorenia imunitnej nedostatočnosti:
Prítomnosť akútneho alebo recidivujúceho (chronického) zápalového infekčného procesu akejkoľvek lokalizácie. Vírusové a/alebo bakteriálne infekcie u novorodencov.
Identifikácia vírusov, oportúnnych baktérií a / alebo húb v ohnisku lézie.
Klinické príznaky charakteristické pre primárne imunodeficiencie u detí.
Prítomnosť dôvodov (imunosupresívne faktory), ktoré spôsobili získané IDD.
Laboratórne príznaky imunodeficiencie.
Na diagnostiku stačia prvé dva znaky, v kombinácii s 3. a 4. alebo bez nich.
Infekčné syndrómy akákoľvek lokalizácia - hlavné klinické "markery" imunodeficiencie a slúžia ako klinické prejavy choroby z imunodeficiencie. Súvislosť s infekciou, „spôsobenou“ oportúnnymi mikroorganizmami (vírusy, baktérie, plesne) s imunodeficienciou je zrejmá, pretože len v jeho prítomnosti je možné ich rozšírenie - infekcia. Práve chýbajúca antivírusová či antibakteriálna imunita vedie k množeniu týchto mikroorganizmov – autológnych alebo zvonku.
Stav odolnosti, imunita tela sú určujúcimi faktormi pri rozvoji akejkoľvek infekcie.
Čo sa týka oportúnnych mikroorganizmov – absolútna väčšina vírusov, baktérií, plesní, rozvoj infekcie s ich účasťou je možný len v imunodeficientnom organizme, t.j. v prítomnosti absolútne a nie relatívna imunodeficiencia niektorého faktora, väzby, receptora alebo molekuly imunity.
Preto bez imunodeficiencie neexistuje žiadna infekcia a je klinickým prejavom IDD. Preto, podobne ako infekcie, má IDD akútny, subakútny a chronický priebeh.
Rozlišovať primárny a sekundárne imunodeficiencie (ID) a podľa toho aj choroby z nedostatku imunity.
Primárne ID - toto je genetické abnormality, zvyčajne sa prejavujú klinicky (aj keď nie vždy!) u detí. Sekundárne ID sa vyskytujú u klinicky zdravých ľudí pod vplyvom rôznych príčin, mnohé z nich však môžu odhaliť genetickú predispozíciu k rozvoju IDD.
Primárne kombinované imunodeficiencie
Ťažké kombinované ID (TKID) .
V tomto stave trpí diferenciácia rôznych buniek, vrátane kmeňových. Existuje niekoľko možností pre SCID.
Ťažká kombinovaná imunodeficiencia s retikulárnou dysgenézou. Mechanizmus: narušená diferenciácia a proliferácia hematopoetických kmeňových buniek na lymfoidné a myeloidné kmeňové bunky. Existuje agranulocytóza, absencia lymfocytov.
Deti zomierajú v prvých mesiacoch života na septický proces.
Ťažká imunodeficiencia s nízkym alebo normálnym počtom B buniek. Mechanizmus a klinický obraz: defekt génu zodpovedného za spoločný γ-reťazec cytokínových receptorov (IL-2, -4, -7) alebo gén proteínkinázy Jak 3; v prvých 6 mesiacoch života dieťaťa začína pretrvávajúca infekcia pľúc, kandidóza hltana, pažeráka, hnačka. Existuje kvantitatívny a / alebo funkčný nedostatok T-buniek, obsah B-buniek môže zodpovedať alebo prekračovať normu, ale tieto bunky vylučujú imunoglobulíny zle, hladiny imunoglobulínov A, M, G sú znížené.
Imunodeficiencia, prejavujúca sa ataxiou-telangiektáziou (Louis-Bar syndróm).
Mechanizmus ID: mutácie, inverzie a translokácie v 7 a 14 chromozómoch, preskupenie génu pre T-receptor a ďalšie zmeny.
Klinika je polymorfná, zmeny v imunitnom systéme v počiatočnej fáze ochorenia sú nevýznamné alebo nie sú pozorované; môžu prevládať neurologické a vaskulárne poruchy, teleangiektázie skléry a kože, cerebelárna ataxia, dysgenéza vaječníkov; ďalšie poškodenie imunitného systému sa zvyšuje; je charakteristický vývoj zdĺhavého, pomalého a chronického zápalu pľúc; smrť na infekčné a vaskulárne neurologické poruchy.
Je znížená hladina T-lymfocytov, vyskytujú sa hladiny IgG, IgG2, IgG4, odpoveď na PHA a bakteriálne antigény, dysimunoglobulinémia, často deficit IgA; niekedy sa vyskytuje hypoplázia týmusu a atrofia lymfatických uzlín, nerovnováha Tx/Tc.
Wiskott-Aldrichov syndróm.
Mechanizmus: defektný gén v Chr11 bol a preto je narušená expresia glykovaného kyslého glykoproteínu sialoporfyrínu (CD43), ktorý sa podieľa na aktivácii T buniek; autozomálne recesívna dedičnosť. Frekvencia je 4: 1 / milión detí.
Klinicky sa prejavuje triádou symptómov – kombináciou ekzému, trombocytopénie a recidivujúcej infekcie.
Existuje lymfocytopénia, T-lymfopénia, znížená hladina T-pomocníkov, trombocytopénia, nie sú žiadne reakcie PCRT, stanovené kožnými testami; znížená odpoveď lymfocytov na PHA a antigény; výrazne znížená hladina IgM, vysoký obsah IgA a IgE, normálna alebo vysoká hladina IgG, znížená tvorba protilátok proti pneumokokovým polysacharidom; makrofágy nerozkladajú polysacharidové antigény.
Klinika: trombocytopénia pri narodení; krvácajúca; ekzém; u detí v prvých mesiacoch života dochádza k opakovaným hnisavým infekciám spôsobeným pneumokokmi a inými baktériami obsahujúcimi polysacharidy; splenomegália; zhubné nádory (5-12%); existuje výrazná hypoplázia týmusovej žľazy a lymfoidného tkaniva.
Imunodeficiencie T-buniek
V týchto podmienkach dochádza k prevládajúcej porážke T-linky imunitného systému.
Aplázia alebo hypoplázia týmusu - DiGeorgeov syndróm.
Mechanizmus: narušený embryonálny vývoj štruktúr 3-4 faryngálnych vačkov, delécia v chromozóme 22q11, nevyvíja sa epitel týmusu a prištítnych teliesok. Chýba funkcia T-buniek; znižuje sa počet lymfocytov a ich funkčná aktivita, zvyšuje sa hladina IgE.
Klinika: aplázia alebo hypoplázia týmusu; malformácie: rázštep podnebia, anomália pravého oblúka aorty, nevyvinutie veľkých ciev, hrudná kosť; katarakta, neonatálna tetánia v dôsledku nedostatočného rozvoja prištítnych teliesok; časté infekčné komplikácie; nie sú žiadne PCZT reakcie; počet lymfocytov v zónach lymfatických uzlín závislých od týmusu je znížený.
Neselofov syndróm .
Je charakterizovaná hypopláziou týmusu, narušením normálneho dozrievania T-lymfocytov, ich nedostatkom v T-dependentných zónach imunitného systému. Funkcie T buniek sú prudko inhibované, celkový počet lymfocytov je znížený, syntéza imunoglobulínov je normálna alebo znížená a produkcia protilátok je inhibovaná.
Nedostatok adenozíndeaminázy (ADA).
Mechanizmus: genetický defekt na lokusu 20. chromozómu - 20.q12 - 13.11, dedičný recesívnym spôsobom; existuje „tichá“ alela lokusu ADA; jeho nedostatok v erytrocytoch a lymfocytoch vedie k hromadeniu deoxyadenozínu, ktorý je toxický pre T-lymfocyty. Už v prvých týždňoch života je zaznamenaná lymfocytopénia; nedostatočnosť T-lymfocytov, objavuje sa hneď po narodení dieťaťa, kombinuje sa s anomáliami vo vývoji skeletu (deformácia, osifikácia), odhaľujú sa známky involúcie týmusu.
B bunkové imunodeficiencie
Pri týchto nedostatkoch je prevažne ovplyvnená B-linka imunitného systému.
Agamaglobulinémia s defektom rastového hormónu spojeným s X chromozómom (Brutonova choroba).
Chlapci sú chorí, pretože v dôsledku mutácie génu Xq22 v dlhom ramene chromozómu X neexistuje tyrozínkináza btkštrukturálne gény na syntézu imunoglobulínov nefungujú. Recesívny typ dedičnosti spojený s chromozómom X. Chýbajúce alebo prudko (menej ako 200 mg / l) znížené hladiny IgM, IgG a IgA; v lymfoidnom tkanive a slizniciach nie sú žiadne plazmatické bunky.
Klinika sa prejavuje vo veku 2 - 3 rokov: odolnosť tela voči baktériám a hubám je znížená a odolnosť voči vírusom je normálna; nie sú žiadne reakcie lymfatických uzlín, sleziny v obdobiach exacerbácií procesu, nedochádza k zvýšeniu adenoidov, hyperplázii mandlí, často v kombinácii s atopickým ekzémom, alergickou rinitídou, bronchiálnou astmou. V súčasnosti pri vykonávaní substitučnej liečby imunoglobulínmi môžu pacienti žiť dostatočne dlho.
Dysimunoglobulinémia .
Ide o selektívny nedostatok jednej alebo viacerých tried imunoglobulínov. Najčastejším z nich je selektívny deficit imunoglobulínu A (1:70-1:100). Táto vada môže prebiehať asymptomaticky, no často sa s ňou spájajú recidívy respiračných a tráviacich chorôb, pretože chráni sliznice pred mikróbmi.
Selektívne deficity IgM alebo IgG sú zriedkavé. Pacienti s nedostatkom IgM zvyčajne zomierajú na sepsu. Nedostatok IgG sa môže prejaviť rôznymi príznakmi v závislosti od chýbajúcich podtried IgG (zvyčajne IgG2). Deficit imunoglobulínov triedy E sa klinicky neprejavuje, existuje však syndróm IgE-hypergamaglobulinémie, ktorý je charakterizovaný rôznymi alergickými prejavmi, ako aj chronickými bakteriálnymi infekciami.
Poruchy mononukleárneho fagocytového a granulocytového systému
Podľa mechanizmu možno takéto ID rozdeliť do štyroch skupín.
Do prvej skupiny patria ID spojené s nedostatočnou aktivitou enzýmov, čo má za následok porucha trávenia absorbovaný predmet.
Do druhej skupiny patrí ID spôsobené porušením chemotaxia fagocyty.
Tretia skupina ID je spojená s nedostatočnosťou opsonizačné faktory sérum (protilátky a komplement).
Štvrtá skupina sa vyznačuje nedostatočnou expresia receptora na povrchu makrofágov (pre C3 zložku komplementu, pre Fc fragmenty Ig atď.).
Napríklad pre nedostatok leukocytových adhezínov (LAD-I syndróm) v dôsledku génového defektu chýba molekula CD18, neadherujú na endotel a nemigrujú do tkaniva.
Chronická granulomatózna choroba charakterizované skutočnosťou, že polynukleárne bunky sú schopné fagocytózy, ale nestrávia absorbované mikróby. Tento proces je založený na defekte NADPH oxidázy, ktorá katalyzuje premenu kyslíka na superoxidový anión, ktorý je nevyhnutný pre prejav baktericídnej aktivity neutrofilov. Vo fagocytoch pretrvávajú katalázo-pozitívne stafylokoky, Klebsiella, Salmonella, Escherichia coli a huby. Vo veku 1-4 rokov sa u detí objavuje ekzémová dermatitída, hnisavé kožné lézie, abscesy v rôznych orgánoch, lymfadenitída, bronchopneumónia, pridružuje sa plesňová infekcia.
Laboratórne diagnostické kritériá sú absencia usmrtenia fagocytovaných baktérií, negatívny a redukovaný NBT test, chemiluminiscencia po fagocytóze zymosanu alebo latexových častíc.
Chédiak-Higashiho syndróm klinicky charakterizované zvýšenou citlivosťou na hnisavé a vírusové infekcie a oslabením farby vlasov, kože a očnej dúhovky. V cytoplazme neutrofilov a makrofágov sa objavujú obrovské granuly, ktoré sa tvoria ako výsledok fúzie cytoplazmatických granúl, ktoré sa detegujú pri farbení na peroxidázu. Súčasne sa pozoruje patologická agregácia melanozómov a v dôsledku toho albinizmus. Zvýšená náchylnosť k infekcii sa vysvetľuje narušením procesu vstupu myeloperoxidázy do vakuol a ich slabou odpoveďou na chemotaktické stimuly.
Nedostatočnosť komplementového systému
V komplementovom systéme možno pozorovať nedostatok ktorejkoľvek zložky a absencia akéhokoľvek faktora blokuje aktiváciu nasledujúcich. To je sprevádzané vývojom rôznych patologických stavov. Nedostatok C1, C2, C4 a C5 sa prejavuje syndrómom podobným systémovému lupus erythematosus. Nedostatok C3 je charakterizovaný opakujúcimi sa hnisavými infekciami.
Okrem deficitu hlavných zložiek existujú deficity inhibítorov komplementového systému: C1-inhibítor a C3-inaktivátor. Klinicky sa prejavuje deficit C1 inhibítora dedičný angioedém ... V dôsledku zvýšenia koncentrácie fragmentu zložky C2, ktorá má vazoaktívny účinok, dochádza k opuchu hrtana, končatín a iných. Typicky sú títo pacienti heterozygotní a syntetizujú malé množstvo inhibítora. Jeho hladinu je možné zvýšiť podávaním anabolických steroidov, prípadne substitučnou liečbou samotným inhibítorom.
Oblasti liečby primárnych imunodeficiencií
Prestup kostná dreň, novorodenecký týmus, embryonálna pečeň - s cieľom nahradiť chýbajúce bunky a vytvoriť podmienky pre ich plnú diferenciáciu. Transplantácia sa používa pri ťažkých kombinovaných ID.
Substitučná liečba imunoglobulínmi, enzýmami, hormónmi týmusu, mediátormi, vitamínmi a inými faktormi.
Antibiotická terapia pri sprievodnej infekcii.
Génová terapia: zavedenie chorých normálnych génov do SI buniek (lymfocytov). Gén adenozíndeaminázy bol prvýkrát zavedený do lymfocytov pacientov s deficitom tohto enzýmu.
Sekundárne imunodeficiencie
Sekundárne (získané) ID sa tvoria pod vplyvom prostredia a sú oveľa bežnejšie ako primárne.
Znaky sekundárnych ID:
nedostatok dedičnej podmienenosti
výskyt na pozadí normálnej reaktivity tela
vzťah s príčinným faktorom, ktorý spôsobil ID
Dôvody sekundárneho ID
1. Nepriaznivé vplyvy prostredia na organizmus a imunitný systém (fyzikálne, chemické, biologické).
2. Choroby ovplyvňujúce imunitný systém:
- vírusové (častejšie)
- alergický a autoalergický, onkologický
- metabolické poruchy, proliferácia buniek, strata bielkovín
- iné závažné ochorenia
3. Imunosupresívna liečba:
- lieková imunosupresia
- žiarenie a iné druhy energie vo veľkých dávkach
- chirurgické zákroky a anestézia
- reakcia štepu proti hostiteľovi (GVHD) po alotransplantácii kostnej drene
4. Fyzický a emocionálny stres
5. Nedostatočná výživa a vyčerpanie (nedostatok bielkovín, tukov-sacharidov, vitamínov, mikroelementov).
6. Profesionálne rizikové faktory (chemické, fyzikálne, psychoemočné).
7. S vekom: predčasnosť detí a patológia starnutia („syndróm starších ľudí“)
Sekundárna, podobne ako primárna, ID môže byť latentná, nemá žiadne klinické príznaky a je zistená iba počas laboratórneho vyšetrenia. ID s klinickými príznakmi je ochorenie imunitnej nedostatočnosti . Klinicky sa prejavuje ako chronické purulentno-zápalové infekcie kože, horných dýchacích ciest, pľúc, urogenitálneho systému, tráviaceho traktu a iných orgánov. Od prechodných (prechodných) posunov v imunitnom systéme sa líšia pretrvávaním porúch v imunitnom systéme po skončení kauzálneho faktora.
Rozlišuje sa závažnosť klinického priebehu pľúca, stredná, subkompenzovaná a ťažká dekompenzovaná sekundárna choroby z imunodeficiencie (imunodeficitné choroby) .
Vírusové ochorenia sekundárnej imunodeficiencie
Vírusy často pretrvávajú v ľudskom tele bez prejavov patológie, t.j. existuje rozšírený nosič vírusov. Týka sa to herpetických vírusov, cytomegalovírusov, adenovírusov, vírusu Epstein-Barrovej a mnohých ďalších. S poklesom hladiny a nedostatkov v interferónovom systéme sú schopné vyvolať imunodeficienciu, a teda VIB niekoľkými spôsobmi:
- transformácia genómu SI buniek;
- priamo ničí imunokompetentné bunky,
- vyvolanie apoptózy;
- väzba na receptory a zmena ich aktivity, chemotaxia, aktivácia supresorov;
- väzbou alebo uvoľnením cytokínov, t.j. modifikácia imunoreaktivity.
Niektoré vírusy majú schopnosť replikovať sa v samotných imunitných bunkách. Príkladom takéhoto mechanizmu môže byť známy tropizmus pre B-lymfocyty vírusu Epstein-Barrovej alebo selektívna porážka T-pomocníkov vírusom HIV. Vírusy mnohých akútne infekcie najmä osýpky, chrípka, rubeola, ovčie kiahne, mumps, herpes - môžu spôsobiť prechodnú anergiu na iné antigény. Klinicky sa prechodná imunosupresia prejavuje vo vývoji vírusových a bakteriálnych komplikácií, ktoré sa často pozorujú pri týchto infekciách. Pretrvávanie vírusov hepatitídy môže viesť k imunomodulácii, potlačeniu T buniek.
Transplacentárne prenosné vírusy (cytomegália, rubeola) majú škodlivý účinok na rôzne tkanivá, vrátane SI buniek. Najvýraznejšie defekty sú opísané pri vrodenej rubeole a cytomegálii. U niektorých detí bola zistená absencia humorálnej a bunkovej imunitnej odpovede na antigény, u iných selektívny deficit IgA. Posledný uvedený defekt sa vysvetľuje schopnosťou vírusu blokovať vývoj buniek v strednom štádiu diferenciácie.
Imunosupresia u detí s aktívnou cytomegalovírusovou infekciou sa prejavuje znížením počtu CD3+, CD4+ T-lymfocytov, inhibíciou fagocytárnej aktivity neutrofilov. Takéto deti sú náchylné na rozvoj bakteriálnych a vírusových superinfekcií.
Poruchy v zložení T- a B-lymfocytov sa pozorujú pri herpetickej infekcii, keď sa počet T-aktivovaných lymfocytov zvyšuje na pozadí všeobecnej T- a B-lymfocytopénie a zníženia expresie molekúl systému HLA. Chronická perzistencia herpetického vírusu v leukocytoch a nervových gangliách vedie k rozvoju ID.
Medzi vírusovou infekciou a deficitom SI existuje komplexný patogenetický vzťah. Vírusová infekcia môže na jednej strane vyvolať sekundárnu imunodeficienciu, na druhej strane sa u pacientov s nedostatočnou imunitou stáva vírusová superinfekcia príčinou ťažkých, život ohrozujúcich stavov, t.j. posilňuje toto ID.
Pretrvávajúce vírusy a intracelulárna imunita
Mnohé vírusy - herpes, cytomegalovírus (CMV), Epstein-Barr, rinovírusy, enterovírusy sú neustále prítomné v bunkách tela a pri pravidelnej aktivácii vyvolávajú rôzne klinické prejavy. Pozoruhodným príkladom sú herpetické vírusy typu 1-8, ktoré perzistujú v nervových gangliách a spôsobujú lézie kože a slizníc, podľa úrovne lokalizácie ganglií - lyabiálne, tarakálne (herpes zoster), sakrálne (genitálne) . Herpes vírusy typu 8 pretrvávajú v T-lymfocytoch, Epstein-Barr - v B-bunkách a iných, CMV - v makrofágoch, leukocytoch, epitelových bunkách. U väčšiny ľudí, ich nosičov, nespôsobujú infekcie, čo je zrejme spôsobené dostatočne vysokou imunitou, predovšetkým interferónovou, pretože nie sú replikované.
Pozoruhodným príkladom vírusom vyvolaného ID je infekcia HIV. Vírus ľudskej imunodeficiencie (HIV) spôsobuje infekčné ochorenie spojené s primárnou léziou SI a rozvojom ťažkej sekundárnej imunodeficiencie, na pozadí ktorej sa aktivuje oportúnna a nepatogénna mikroflóra.
Sekundárne imunodeficiencie pri chorobách
Všetky závažné ochorenia vedú k rozvoju imunologickej nedostatočnosti.
Metabolické poruchy sú jednou z príčin sekundárnej imunodeficiencie. Pri diabetes mellitus je napríklad inhibovaná chemotaxia a fagocytárna aktivita neutrofilov, čo vedie k kožnej pyodermii a abscesom.
S popáleninami ID vznikajú v súvislosti s veľkou stratou imunoglobulínov a komplementových proteínov z krvnej plazmy. Ak plocha kožných lézií presiahne 30 %, vznikajú poruchy bunkovej imunity.
Nádory vylučujú imunomodulačné faktory a cytokíny, ktoré potláčajú imunitu. Dochádza k zníženiu počtu T-lymfocytov, zvýšeniu aktivity supresorových buniek, inhibícii fagocytózy. Obzvlášť výrazné zmeny sa vyskytujú pri bežných nádorových procesoch s metastázami.
Sekundárne imunodeficiencie pri rôznych patofyziologických stavoch a strese
Pri chronickom hladovaní dochádza k imunodeficienciám v dôsledku nedostatku bielkovín, vitamínov a mikroelementov. V týchto prípadoch trpí predovšetkým bunkový imunitný systém: znižuje sa odpoveď lymfocytov na mitogény, pozoruje sa atrofia lymfoidného tkaniva a je narušená funkcia neutrofilov.
Ťažká fyzická aktivita a sprievodný stres u profesionálnych športovcov v závislosti od trvania záťaže spôsobujú dočasnú alebo pretrvávajúcu imunomoduláciu. Dochádza k poklesu hladiny imunoglobulínov, subpopulácií T-lymfocytov, k aktivite fagocytózy. Počas tohto „obdobia imunodeficiencie“ sú športovci vysoko náchylní na rôzne infekcie. Hodnoty CI sa zvyčajne vrátia do normálu počas odpočinku, ale nie vždy.
Sekundárne ID pre chirurgické operácie sú spojené so silnou stresovou reakciou a s pôsobením liekov na anestéziu. Vzniká dočasný imunodeficitný stav, pri ktorom klesá počet T- a B-lymfocytov, znižuje sa ich funkčná aktivita. Porušené indikátory sa obnovia až po mesiaci, ak neexistujú žiadne faktory, ktoré potláčajú imunitu.
So starnutím organizmu, ID sú výsledkom imunomodulácií vznikajúcich vplyvom nepriaznivých faktorov a z chorôb, najmä vírusových. U zdravých starších ľudí (90-100 rokov) sú ukazovatele SI blízke hodnotám u ľudí stredného veku, hoci majú svoje vlastné charakteristiky.
Novorodenec a malé deti majú iné ukazovatele SI ako dospelí; cirkulujú materské IgG získané cez placentu, ktorého hladina klesá v 3-6 mesiaci, čo nie je ID. Predčasne narodené deti sa rodia s rôznymi defektmi SI spojenými s jeho nezrelosťou a často aj s vnútromaternicovými infekciami. Umelé kŕmenie detí spôsobuje deficit sekrečného IgA a ďalších ochranných faktorov (lyzozým a pod.) materského mlieka.
Imunodeficiencia je porušením ochranných funkcií ľudského tela v dôsledku oslabenia imunitnej odpovede na patogény rôzneho charakteru. Veda opísala celý rad druhov tohto druhu. Táto skupina ochorení je charakterizovaná nárastom a zhoršením priebehu infekčných ochorení. Zlyhania v práci imunity sú v tomto prípade spojené so zmenou kvantitatívnych alebo kvalitatívnych charakteristík jej jednotlivých zložiek.
Vlastnosti imunity
Imunitný systém zohráva podstatnú úlohu v normálnom fungovaní tela, pretože je určený na detekciu a ničenie antigénov, ktoré môžu prenikať z vonkajšieho prostredia (infekčné) a byť dôsledkom nádorového rastu vlastných buniek (endogénne). Ochrannú funkciu zabezpečujú predovšetkým vrodené faktory ako fagocytóza a komplementový systém. Získané a bunkové reakcie sú zodpovedné za adaptačnú reakciu organizmu. Spojenie celého systému nastáva prostredníctvom špeciálnych látok - cytokínov.
V závislosti od príčiny vzniku sa stav poruchy imunity delí na primárne a sekundárne imunodeficiencie.
Čo je primárna imunodeficiencia
Primárne imunodeficiencie (PID) sú poruchy imunitnej odpovede spôsobené genetickými defektmi. Vo väčšine prípadov sú zdedené a ide o vrodené abnormality. PID sa najčastejšie vyskytujú v ranom veku, ale niekedy nie sú diagnostikované až do dospievania alebo dokonca dospelosti.
PID – skupina vrodené choroby, sa líšili v klinických prejavoch. Medzinárodná klasifikácia chorôb zahŕňa 36 opísaných a dostatočne preštudovaných stavov primárnej imunodeficiencie, no podľa lekárskej literatúry ich je okolo 80. Faktom je, že nie u všetkých chorôb boli identifikované zodpovedné gény.
Len génové zloženie chromozómu X sa vyznačuje minimálne šiestimi rôznymi imunodeficienciami, a preto je výskyt takýchto ochorení u chlapcov rádovo vyšší ako u dievčat. Existuje predpoklad, že vnútromaternicová infekcia môže mať etiologický vplyv na vznik vrodenej imunodeficiencie, toto tvrdenie však zatiaľ nie je vedecky potvrdené.

Klinický obraz
Klinické prejavy primárnych imunodeficiencií sú rovnako rôznorodé ako samotné stavy, ale existuje jeden spoločný príznak – hypertrofovaný infekčný (bakteriálny) syndróm.
Imunodeficiencie, primárne aj sekundárne, sa prejavujú sklonom pacientov k častým recidivujúcim (recidivujúcim) ochoreniam infekčnej etiológie, ktoré môžu byť spôsobené atypickými patogénmi.
Najčastejšie sú týmito ochoreniami postihnutý bronchopulmonálny systém a orgány ORL človeka. Často sú postihnuté aj sliznice a koža, čo sa môže prejaviť abscesmi a sepsou. Bakteriálne patogény spôsobujú bronchitídu a sinusitídu. Ľudia s oslabenou imunitou často pociťujú skorú plešatosť a ekzém a niekedy aj alergické reakcie. Časté sú aj autoimunitné poruchy a sklon k zhubným novotvarom. Imunodeficiencia u detí takmer vždy spôsobuje oneskorenie duševného a fyzického vývoja.
Mechanizmus vývoja primárnych imunodeficiencií
Klasifikácia chorôb podľa mechanizmu ich vývoja je najinformatívnejšia v prípade štúdia stavov imunodeficiencie.

Lekári rozdeľujú všetky choroby imunitnej povahy do 4 hlavných skupín:
Humorálne alebo B-bunky, medzi ktoré patrí Brutonov syndróm (agamaglobulinémia spojená s chromozómom X), nedostatok IgA alebo IgG, nadbytok IgM pri všeobecnom nedostatku imunoglobulínu, jednoduchá variabilná imunodeficiencia, prechodná hypogamaglobulinémia novorodencov a množstvo ďalších ochorení spojených s humorálnou imunitou.
Primárne imunodeficiencie T-buniek, ktoré sa často nazývajú kombinované, pretože pri prvých poruchách je vždy narušená humorálna imunita, napríklad hypoplázia (Di Georgeov syndróm) alebo dysplázia (T-lymfopénia) týmusu.
Imunodeficiencie spôsobené poruchami fagocytózy.
Imunodeficiencie v dôsledku zhoršenej práce
Náchylnosť na infekcie
Pretože príčinou imunodeficiencie môže byť porušenie rôznych väzieb
imunitný systém, potom citlivosť na infekčné agens nebude pre každý konkrétny prípad rovnaká. Takže napríklad pri humorálnych ochoreniach je pacient náchylný na infekcie, ktoré sú spôsobené streptokokmi, stafylokokmi, pričom tieto mikroorganizmy často vykazujú rezistenciu na antibakteriálne lieky. Pri kombinovaných formách imunodeficiencie sa na baktérie môžu uchytiť vírusy, ako sú herpes alebo plesne, ktoré sú reprezentované najmä kandidózou. Fagocytárna forma je charakterizovaná hlavne rovnakými stafylokokmi a gramnegatívnymi baktériami.

Prevalencia primárnych imunodeficiencií
Dedičné imunodeficiencie sú pomerne zriedkavé ľudské ochorenia. Frekvencia výskytu takýchto porúch imunity sa musí posudzovať vo vzťahu ku každému konkrétnemu ochoreniu, keďže ich prevalencia nie je rovnaká.
Vrodenou dedičnou imunodeficienciou bude trpieť v priemere len jeden novorodenec z päťdesiatich tisíc. Najčastejším ochorením v tejto skupine je selektívny deficit IgA. Vrodená imunodeficiencia tohto typu sa vyskytuje v priemere u jedného z tisíc novorodencov. Navyše 70 % všetkých prípadov nedostatku IgA súvisí s úplnou nedostatočnosťou tejto zložky. Zároveň sa niektoré zriedkavejšie choroby človeka imunitného charakteru, dedičné, môžu šíriť v pomere 1: 1 000 000.
Ak vezmeme do úvahy frekvenciu výskytu ochorení PID v závislosti od mechanizmu, potom existuje veľmi zaujímavý obrázok... Primárne imunodeficiencie B-buniek, alebo, ako sa im tiež hovorí, poruchy tvorby protilátok, sa vyskytujú častejšie ako iné a predstavujú 50 – 60 % všetkých prípadov. Súčasne sú T-bunkové a fagocytárne formy diagnostikované u 10-30% pacientov. Najvzácnejšie sú ochorenia imunitného systému spôsobené defektmi komplementu - 1-6%.
Treba tiež poznamenať, že údaje o frekvencii výskytu PID sú v rôznych krajinách veľmi rozdielne, čo môže súvisieť s genetickou predispozíciou určitej národnej skupiny k určitým mutáciám DNA.
Diagnóza imunodeficiencie
Primárna imunodeficiencia u detí je najčastejšie určená mimo čas, v dôsledku
s tym, ze na urovni obvodneho pediatra je dost tazke urobit takuto diagnozu.

To spravidla vedie k oneskorenému začiatku liečby a nepriaznivej prognóze terapie. Ak lekár na základe klinického obrazu ochorenia a výsledkov celkových vyšetrení navrhol stav imunodeficiencie, v prvom rade by mal poslať dieťa na konzultáciu k imunológovi.
V Európe existuje Asociácia imunológov, ktorá sa zaoberá štúdiom a vývojom metód liečby tohto druhu ochorení, nazývaná EOI (European Society for Immunodeficiencies). Vytvorili a neustále aktualizujú databázu PID ochorení a schválili diagnostický algoritmus pre pomerne rýchlu diagnostiku.
Diagnóza začína odobratím anamnézy choroby. Osobitná pozornosť by sa mala venovať genealogickému aspektu, pretože väčšina vrodených imunodeficiencií je dedičná. Ďalej, po fyzickom vyšetrení a získaní údajov zo všeobecných klinických štúdií sa vykoná predbežná diagnóza. V budúcnosti, aby sa potvrdila alebo vyvrátila domnienka lekára, musí pacient absolvovať dôkladné vyšetrenie u špecialistov, akými sú genetik a imunológ. Až po vykonaní všetkých vyššie opísaných manipulácií môžeme hovoriť o konečnej diagnóze.
Laboratórny výskum

Ak počas diagnózy existuje podozrenie na syndróm primárna imunodeficiencia mali by sa vykonať nasledujúce laboratórne testy:
Stanovenie podrobného krvného obrazu (osobitná pozornosť sa venuje počtu lymfocytov);
Stanovenie obsahu imunoglobulínov v krvnom sére;
Kvantifikácia B- a T-lymfocytov.
Dodatočný výskum
Okrem laboratórnych diagnostických testov, ktoré sú už uvedené vyššie, sa v každom prípade priradia individuálne dodatočné testy. Existujú rizikové skupiny, ktoré musia byť testované na infekciu HIV alebo genetické abnormality. Lekár tiež počíta s možnosťou, že existuje ľudská imunodeficiencia 3 alebo 4 typov, pri ktorej bude trvať na podrobnom štúdiu fagocytózy pacienta nastavením testu s indikátorom tetrazolínovej modrej a kontrolou zloženia komponentov komplementového systému. .
Liečba PID
Je zrejmé, že potrebná terapia bude závisieť predovšetkým od samotného imunitného ochorenia, ale vrodenú formu, žiaľ, nemožno úplne odstrániť, čo sa nedá povedať o získanej imunodeficiencii. Na základe moderného medicínskeho vývoja sa vedci snažia nájsť spôsob, ako odstrániť príčinu na genetickej úrovni. Aj keď ich pokusy neboli korunované úspechom, možno konštatovať, že imunodeficiencia je nevyliečiteľný stav. Pozrime sa na princípy aplikovanej terapie.

Substitučná terapia
Liečba imunodeficiencie sa zvyčajne znižuje na substitučnú liečbu. Ako už bolo spomenuté vyššie, telo pacienta nie je schopné samostatne produkovať niektoré zložky imunitného systému, alebo je ich kvalita výrazne nižšia, ako je potrebné. Terapia bude v tomto prípade spočívať v medikácii protilátok alebo imunoglobulínov, ktorých prirodzená tvorba je narušená. Najčastejšie sa lieky podávajú intravenózne, ale niekedy je možná aj subkutánna cesta, aby sa uľahčil život pacienta, ktorý v tomto prípade nemusí znova navštevovať zdravotnícke zariadenie.
Princíp substitúcie často umožňuje pacientom viesť takmer normálny život: štúdium, prácu a odpočinok. Ochorením oslabená imunita, humorálne a bunkové faktory a neustála potreba podávania drahých liekov pacientovi samozrejme neumožnia úplne sa uvoľniť, ale stále je to lepšie ako život v tlakovej komore.
a prevencia
Vzhľadom na to, že každá bakteriálna alebo vírusová infekcia, pre zdravého človeka bezvýznamná, pre pacienta s ochorením zo skupiny primárnej imunodeficiencie môže byť smrteľná, je potrebné správne vykonávať prevenciu. Tu prichádzajú na rad antibakteriálne, protiplesňové a antivírusové lieky. malo by sa to robiť práve na preventívnych opatreniach, pretože oslabený imunitný systém nemusí dovoliť poskytnúť kvalitnú liečbu.
Okrem toho treba pamätať na to, že takíto pacienti sú náchylní na alergické, autoimunitné a ešte horšie na neoplastické stavy. To všetko bez úplného lekárskeho dohľadu nemusí človeku umožniť viesť plnohodnotný životný štýl.
Transplantácia
Keď sa odborníci rozhodnú, že pacient nemá inú možnosť chirurgický zákrok, možno vykonať transplantáciu kostnej drene. Tento postup je spojená s viacerými rizikami pre život a zdravie pacienta a v praxi ani v prípade úspešného výsledku nedokáže vždy vyriešiť všetky problémy poruchy imunity. Pri vykonávaní takejto operácie sa celý príjemca nahradí tým istým, ktorý poskytol darca.
Primárne imunodeficiencie sú najťažším problémom modernej medicíny, ktorý, žiaľ, ešte nie je úplne vyriešený. Pri ochoreniach tohto druhu stále prevláda nepriaznivá prognóza, čo je dvojnásobne smutné vzhľadom na skutočnosť, že najčastejšie ich postihujú deti. Mnohé formy imunitnej nedostatočnosti sú však zlučiteľné s plnohodnotným životom za predpokladu, že sú včas diagnostikované a je aplikovaná adekvátna liečba.
Ľudský imunitný systém je navrhnutý tak, aby včas reagoval na inváziu cudzích prvkov. Jeho správnou funkcionalitou je rozpoznať hrozbu a odstrániť ju. Primárna imunodeficiencia znamená, že dieťa si počas vnútromaternicového vývoja nevytvorilo ochranný mechanizmus, alebo ho nedostalo v dôsledku dedičného faktora. Výsledkom je, že škodlivé mikroorganizmy, ktoré sa dostanú do jeho tela, mu maximálne ublížia. To isté možno povedať o atypických bunkách, ktoré majú negatívny vplyv na zdravie a spôsobujú patológie rôznej závažnosti.
Je potrebné rozlišovať medzi primárnou a sekundárne imunodeficiencie... Primárna sa určuje u dojčaťa krátko po narodení. Jeho telo je zbavené schopnosti brániť sa proti antigénom, je náchylné na infekčnú inváziu. To sa prejavuje tým, že dieťa je často choré, trpí opakovanými chorobami, ťažko ich znáša a má komplikácie. Ťažké formy primárnej imunodeficiencie vedú k smrti v detstve.
Existujú zriedkavé prípady, keď sa primárna imunitná nedostatočnosť prejavila u dospelých. To je možné, ale na to musí mať človek vysokú kompenzáciu za určitý druh ochorenia.
Klinika choroby je opätovná infekcia, prechod chorôb na chronická forma... Čo vedie k primárnej imunodeficiencii:
- Pacient trpí bronchopulmonálnymi abnormalitami.
- Postihnuté sú jeho sliznice a koža.
- Existujú problémy s orgánmi ORL.
- PIDS spravidla vedie k lymfadenitíde, abscesom, osteomyelitíde, meningitíde, sepse.
- Niektoré formy primárnej imunodeficiencie vyvolávajú alergie, autoimunitné ochorenia a rast malígnych novotvarov.
Imunológia sa zaoberá štúdiom dysfunkcií imunitnej obrany – veda o vývoji a tvorbe obranného mechanizmu, ktorý pôsobí proti prenikaniu antigénov do organizmu a ničí bunky poškodené škodlivými látkami a mikroorganizmami.
Čím skôr sa PIDS diagnostikuje, tým väčšiu šancu má dieťa na prežitie a pokračovanie v živote v uspokojivom zdraví. Včasné rozhodnutie je nevyhnutné génová mutácia, ktorá umožňuje rozhodovať o plánovaní rodičovstva.
Imunodeficiencia je považovaná za pretrvávajúcu anomáliu obranného mechanizmu, ktorá spôsobuje zlyhanie imunitnej odpovede na vplyv antigénov. Toto zlyhanie môže byť štyroch typov:
- vek, to znamená, že vzniká v detstve alebo v starobe;
- získané kvôli podvýživaživotný štýl, lieky, vírus AIDS atď.;
- vyvinuté v dôsledku rôznych infekcií;
- vrodené alebo primárne ID.
PIDS sú klasifikované podľa formy a závažnosti ochorenia. Primárne imunodeficiencie zahŕňajú:
- ID charakterizované porážkou niekoľkých bunkových komplexov;
- Retikulárna dysgenéza, pri ktorej chýbajú kmeňové bunky, odsúdi novorodenca na smrť.
- Ťažká kombinovaná ID je dedičné ochorenie spôsobené dysfunkciou B a T lymfocytov.
- DiGeorgeov syndróm - alebo abnormality týmusu, prištítnych teliesok - nedostatočný rozvoj alebo absencia týmusu. Následkom defektu dochádza k postihnutiu T-lymfocytov, vrodeným srdcovým chybám, deformáciám stavby kostí, stavby tvárových kostí, obličkovým defektom a poruchám funkcie centrálneho nervového systému.
- Primárna imunodeficiencia v dôsledku poškodenia B-lymfocytov.
- Poruchy v myeloidných bunkách, ktoré vyvolávajú chronické granulomatózne ochorenie (CGD) s abnormalitou v metabolizme kyslíka. Porucha tvorby aktívneho kyslíka vedie k chronickým plesňovým a bakteriálnym infekciám.
- Defekty komplexných krvných bielkovín, ktoré zhoršujú humorálnu ochranu. V komplementovom systéme môže chýbať niekoľko komponentov.
Potreba vedieť! Bunková imunodeficiencia je charakterizovaná nedostatkom imunokompetentných buniek, medzi ktoré patria lymfocyty, plazmatické bunky, makrofágy. Humorálna imunodeficiencia znamená dysfunkciu tvorby protilátok.
Príznaky primárnej imunodeficiencie
Príznaky a symptómy naznačujú primárne imunodeficiencie. Pri štúdiu klinického obrazu priebehu ochorenia lekári kliniky identifikujú typ imunitnej nedostatočnosti. Toto je uľahčené vyšetrením, analýzami, zberom anamnézy na objasnenie genetickej patológie.

- Primárne nedostatky bunkovej imunity spôsobujú vírusové a plesňové infekcie. Za charakteristické znaky sa považujú opakované prechladnutia, ťažké ARVI, ovčie kiahne, mumps, časté prejavy herpesu. Pacient trpí drozdom, zápalom pľúc, infekciami gastrointestinálneho traktu spôsobenými hubami. Bunková imunodeficiencia zvyšuje riziko rakoviny a lymfómu.
- Nedostatok humorálnej ochrany vyvoláva bakteriálne infekcie. Ide o zápal pľúc, kožné abscesy, erysipel, stafylokoky, streptokoky.
- Nedostatočná hladina sekrečného imunoglobulínu A spôsobuje poškodenie slizníc v ústach, nose, očiach, črevách, prieduškách trpia.
- Kombinovaná ID je charakterizovaná komplikáciami vírusových a bakteriálne infekcie... Prejavy tejto formy primárnej imunodeficiencie sú nešpecifické - prejavujú sa v malformáciách, nádorových procesoch, lymfoidných tkanivách, týmusu, megaloblastickej anémii.
- Vrodená neutropénia a dysfunkcia fagocytózy granulocytov vedie k bakteriálnym zápalovým procesom s abscesmi, abscesmi. Výsledkom môže byť sepsa.
- Primárne imunodeficiencie spojené s komplementom vedú k bakteriálnym infekciám, autoimunitným ochoreniam, ako aj k opakujúcim sa edémom na tele, končatinách - dedičný angioedém (HAE).
Príčiny primárnej imunodeficiencie
Dysfunkcie imunitného systému sa tvoria v embryu vo vnútri maternice. Tento proces je ovplyvnený rôznymi faktormi. Prenatálna diagnostika ukazuje kombináciu vrodené chyby vývoj plodu s imunodeficienciou. Etiológia PIDS je založená na troch patológiách.
- Genetické mutácie, čo znamená, že zmeny nastali v génoch, na ktorých imunokompetentné bunky vykonávajú svoje funkcie. To znamená, že proces vývoja a diferenciácie buniek je narušený. Dedičnosť anomálie je autozomálne recesívna, keď obaja rodičia sú nosičmi mutagénu. Len malý počet mutácií sa vyvinie spontánne alebo zárodočnou líniou (v zárodočných bunkách).
- Teratogénnym faktorom je vplyv nebezpečných toxínov na embryo, ktoré vedú k vrodenej primárnej imunodeficiencii. Vyvolávajú sa infekcie ID TORCH - cytomegalovírus, herpes, rubeola, toxoplazmóza u tehotných žien.
- Nejasná etiológia. Imunitný deficit, ktorého príčina nie je jasná.
Takéto stavy zahŕňajú asymptomatické ID, ktoré sa prejavujú infekčnými komplikáciami v provokačných situáciách. Ak je čo i len jeden z prvkov obranného mechanizmu vystavený anomáliám, tak obrana oslabuje, pacient sa stáva objektom pre inváziu rôznych infekcií.
Diagnóza primárnej imunitnej nedostatočnosti
Stavy imunodeficiencie sa identifikujú podľa typu, keďže primárny ID je najčastejšie vrodený, potom sa jeho typ určuje v prvých mesiacoch alebo týždňoch. Návšteva lekára je potrebná pri častých ochoreniach dieťaťa, prechladnutí, vzniku plesňových, vírusových, bakteriálnych infekcií. Anomálie vo vývoji dieťaťa môžu závisieť aj od primárnej imunodeficiencie. Na vyriešenie problému je potrebná urgentná diagnóza a okamžité začatie liečby.

Metóda identifikácie choroby zahŕňa nasledujúce postupy:
- všeobecné vyšetrenie, pri ktorom sa pozornosť venuje léziám kože, slizníc, pustulóznym procesom, podkožnému edému tukového tkaniva;
- štúdium leukocytového vzorca podľa všeobecná analýza krv, ID je indikovaná prítomnosťou leukopénie, neutropénie, agranulocytózy a iných porúch;
- biochemické vyšetrenie krvi ukazuje dysgamaglobulinémiu, prítomnosť necharakteristických metabolitov, čo naznačuje primárny humorálny ID;
- špecifický výskum reakcie imunitného systému. Študujú sa ukazovatele aktivity imunokompetentných buniek;
- molekulárno-genetická analýza - metóda sekvenovania génov pre typ mutácie. Toto je spôsob, ako definovať syndrómy Bruton, Dee Giorgi, Duncan, Wiskott-Aldrich.
Lekár rozlišuje stavy imunodeficiencie so získaným sekundárnym ID, vznikajúce vplyvom žiarenia, toxických látok, autoimunitných ochorení a onkológie. U dospelých je diagnostika ťažká, pretože príznaky sú vyhladené, príznaky sú implicitné.
Prenatálna diagnostika
Stanovenie primárneho ID biopsiou choriových klkov sa nazýva prenatálna identifikácia formy ochorenia. Okrem toho sa študuje kultúra buniek plodových vôd, fetálna krv. Ide o komplexné testy, ktoré sa zobrazujú v prípadoch, keď sa u rodičov zistí mutagén.
Ale na identifikáciu X-viazaného ťažkého kombinovaná imunodeficiencia táto metóda poskytuje presný výsledok a tiež objasňuje diagnózu pri primárnych ID syndrómoch, chronickej granulomatóze a iných SCID stavoch.
Liečba primárnych imunodeficiencií
Rôzna etiológia a patogenéza chorôb neumožňuje vyvinúť všeobecnú metodiku liečby patológie. Pri ťažkých formách nie je terapeutická liečba relevantná, prináša len dočasnú úľavu, ale smrť je nevyhnutná na komplikácie imunodeficiencie. V týchto prípadoch pomáha iba transplantácia kostnej drene alebo embryonálneho týmusu.
Nedostatok bunkovej imunity je kompenzovaný použitím špecifických liečiv stimulujúcich kolónie. Ide o substitučnú imunoterapiu tymalínom, taktivínom, levamizolom a inými prostriedkami, ktorých výber robí imunológ. Fermentopatie sú korigované enzýmami, metabolitmi. Biotín je bežný liek v tejto sérii.

Dysglobulinémia (nedostatok humorálnej ochrany) sa lieči náhradou imunoglobulínu v závislosti od chýbajúcich látok tohto typu. Ale hlavnou prekážkou progresie ochorenia je prevencia infekcií. Navyše očkovanie detí s primárnym ID nemá žiadny účinok, je nebezpečné.
Prognóza a prevencia
S ťažkým primárnym ID je dieťa odsúdené na zánik, zomiera v prvom roku života. Ďalšie patológie imunitného systému sa liečia tak, ako je opísané vyššie. Hlavnou úlohou rodičov je včasná návšteva lekára a starostlivosť o deti. Dieťa by nemalo byť infikované vírusovými, bakteriálnymi, hubovými patogénmi.
Ak plánujete bábätko a máte problémy s génovou mutáciou, potom je potrebná konzultácia s imunológom. Počas tehotenstva musíte absolvovať prenatálnu diagnostiku, chrániť sa pred infekciami a dodržiavať všetky odporúčania lekára.
U pacientov s ID je dôležité dodržiavať osobnú hygienu, starostlivo sa starať o ústnu dutinu, sliznicu nosa, oči bez poškodenia ich celistvosti. Potrebné vyvážená strava, vylúčenie kontaktu s pacientmi počas epidémií, drogovej prevencie infekcií.
Komplikácie po imunodeficiencii
Primárne imunodeficiencie vedú k hrozivým komplikáciám. Výsledkom následkov môže byť smrť človeka. Tieto stavy sa považujú za sepsu, abscesy, zápal pľúc, závažné infekcie. Autoimunitné ochorenia sú možné, keď zlyhanie imunitného systému spočíva v tom, že ničí svoje vlastné bunky. Zvyšuje sa riziko rakoviny a nerovnováhy gastrointestinálneho traktu, kardiovaskulárneho systému.
Záver
Primárna imunodeficiencia nie je vždy veta. Je potrebné neustále sledovať imunológ, pomôže to udržať uspokojivú kvalitu života a dlho žiť.
Imunodeficiencia je zníženie kvantitatívnych ukazovateľov a / alebo funkčnej aktivity hlavných zložiek imunitného systému, čo vedie k narušeniu obranyschopnosti tela proti patogénnym mikroorganizmom a prejavuje sa zvýšenou infekčnou chorobnosťou.
Ako viete, hlavnou funkciou imunitného systému je rozpoznávanie a eliminácia cudzorodých látok antigénneho charakteru, ktoré prenikajú do tela z prostredia (mikroorganizmy) alebo vznikajú endogénne (nádorové bunky). Táto funkcia je realizovaná pomocou faktorov vrodenej imunity (fagocytóza, antimikrobiálne peptidy, proteíny komplementového systému, NK-bunkového systému a pod.) a získanej, resp. adaptívnej imunity, uskutočňovanej pomocou bunkových a humorálnych imunitných reakcií. K regulácii aktivity zložiek imunitnej obrany organizmu a ich interakcii dochádza pomocou cytokínov a medzibunkových kontaktov.
V každej z uvedených zložiek imunitného systému, ako aj v mechanizmoch ich regulácie, môžu nastať poruchy vedúce k rozvoju imunodeficiencie, ktorej hlavným klinickým prejavom je precitlivenosť na patogény infekčných ochorení. Existujú 2 typy imunodeficiencie: primárne a sekundárne.
Primárne imunodeficiencie(PID) - dedičné ochorenia spôsobené defektmi v génoch, ktoré riadia imunitnú odpoveď. PID - choroby rôzneho charakteru a závažnosti porúch imunity, klinických prejavov a molekulárnych porúch. Klinický obraz PID je charakterizovaný opakovanými a chronickými, závažnými infekčnými procesmi, najmä bronchopulmonálneho systému.
a orgánov ENT, kože a slizníc; môže sa vyvinúť hnisavá lymfadenitída, abscesy, osteomyelitída, meningitída a sepsa. V niektorých formách dochádza k prejavom alergií, autoimunitných ochorení a vzniku niektorých zhubné nádory... Pozornosť by sa mala venovať oneskoreniu z hľadiska vekových ukazovateľov fyzického vývoja. V súčasnosti je popísaných asi 80 PID a boli identifikované gény zodpovedné za vznik väčšiny týchto ochorení. Adekvátne laboratórne testy umožňujú odlíšiť patológiu na úrovni lymfocytov a patológiu na úrovni nelymfocytárnych mechanizmov deštrukcie a eliminácie antigénov.
Prevalencia PID závisí od formy ochorenia a v priemere sa pohybuje od 1:10 000 do 1:100 000 novorodencov. Napríklad selektívny nedostatok IgA je oveľa bežnejší od 1:500 do 1:1500 ľudí vo všeobecnej populácii. Prevalencia rôznych foriem PID sa v jednotlivých krajinách líši. Najčastejšie poruchy tvorby protilátok - 50-60% prípadov, kombinované PID - 10-30%, defekty fagocytózy - 10-20%, defekty komplementu - 1-6%. Väčšina PID sa prejavuje v ranom detstve, hoci neskorší nástup niektorých foriem PID, najmä bežného variabilného imunologického deficitu (CVID), je možný.
Podľa mechanizmov vývoja existujú 4 hlavné skupiny PID:
1. skupina - hlavne humorálna, alebo B-bunková
PID;
Skupina 2 - kombinované PID (so všetkými imunodeficienciami T-buniek existuje dysfunkcia B-buniek);
3. skupina - PID spôsobené defektmi pri fagocytóze;
4. skupina - PID, spôsobené poruchami systému komplementu.
Zásady diagnostiky primárnych imunodeficiencií
Včasná diagnostika a včasné začatie liečby určujú prognózu ochorenia. Stanovenie diagnózy na úrovni obvodných pediatrov predstavuje určité ťažkosti, ktoré sú často dôsledkom chýbajúcej možnosti včasnej konzultácie pacienta imunológom a špeciálneho laboratórneho imunologického vyšetrenia (tab. 11-1). Hoci znalosť znakov klinického obrazu PID a zmien
vo všeobecnej klinickej laboratórne analýzy umožniť podozrenie na PID a poslať pacienta k špecialistovi. Európska spoločnosť pre imunodeficiencie vyvinula protokoly na včasnú diagnostiku PID a vytvorila aj elektronickú databázu Európskeho registra PID. Algoritmus diagnostiky PID je znázornený na obr. 11-1.
Tabuľka 11-1. Etapy imunologického vyšetrenia pri podozrení na imunodeficienciu
Ryža. 11-1. Algoritmus na diagnostiku primárnej imunodeficiencie
Všeobecné znaky klinického obrazu primárnej imunodeficiencie
Vedenie v klinický obraz PID je takzvaný infekčný syndróm - zvýšená náchylnosť na patogény infekčných chorôb všeobecne, neobvykle ťažké recidivujúce (recidivujúce) klinický priebeh, prítomnosť atypických patogénov (často oportúnnych) v etiológii ochorenia. Typ patogénu je určený povahou imunitného defektu. Pri poruchách tvorby protilátok je možné identifikovať flóru rezistentnú na antibakteriálne lieky - stafylokoky, streptokoky, Haemophilus influenzae. Pri imunitnej nedostatočnosti T-buniek sa okrem baktérií zisťujú vírusy (napríklad rodina herpesvírusov), huby (Candida spp., Aspergillus a ďalšie) a s fagocytárnymi defektmi - stafylokoky, gramnegatívne baktérie, huby atď.
Laboratórny výskum
Ak klinické dôkazy naznačujú PID, mali by sa vykonať nasledujúce štúdie:
Stanovenie podrobného krvného obrazu (dôležité sú najmä kvantitatívne a percentuálne ukazovatele lymfocytov);
Stanovenie hladín IgG, IgA a IgM v krvnom sére;
Počítanie subpopulácií T- a B-lymfocytov;
Pre špeciálne indikácie:
◊ analýza funkčného stavu fagocytov (najjednoduchšia a najinformatívnejšia analýza je test na obnovenie tetrazóliovej modrej);
◊ analýza obsahu hlavných zložiek komplementu (začnite s C3 a C4);
◊ analýza na infekciu HIV (ak existujú možné rizikové faktory);
◊ molekulárne genetické štúdie, ak sú indikované.
Princípy liečby primárnych imunodeficiencií
Hlavným cieľom terapie PID je liečba komplikácií ochorenia a ich prevencia. Tento prístup je spôsobený tým, že defekty imunitného systému pri PID sú na genetickej úrovni. V súčasnosti prebieha intenzívny výskum gen
terapia imunodeficiencií, ktoré môžu viesť k vzniku ďalších radikálne metódy ich liečbe.
V závislosti od formy PID liečba pozostáva zo substitučnej terapie, liečby a prevencie infekčných, autoimunitných prejavov ochorenia, liečby malígnych novotvarov a použitia špeciálnych metód vrátane transplantácie krvotvorných buniek (v závislosti od typu PID).
PORUCHY IMUNOGLOBULÍNU
Prechodná hypogamaglobulinémia u detí
Prechodná hypogamaglobulinémia u detí je spojená s fyziologickým znakom fázovej tvorby imunoglobulínového systému. Dozrievanie tvorby IgM a IgA protilátok je „oneskorené“ v najväčšej miere. U zdravých detí sa obsah materského IgG postupne znižuje a po šiestich mesiacoch sa zvyšuje tvorba vlastných IgG protilátok. U niektorých detí je však vzostup hladín imunoglobulínov oneskorený. Takéto deti môžu trpieť opakovanými bakteriálnymi infekčnými ochoreniami. V týchto prípadoch by sa nemali používať infúzie darcovského imunoglobulínu (intravenózny imunoglobulín).
Selektívny nedostatok imunoglobulínu A
Selektívny deficit imunoglobulínu A (SD IgA - Selektívny nedostatok IgA) sa vyvíja v dôsledku génového defektu tnfrsf13b
alebo p). Deficit IgA v prítomnosti imunoglobulínov iných tried je najčastejšou imunodeficienciou zistenou v bežnej populácii s frekvenciou 1: 500-1500 osôb (ešte častejšie u alergikov). Rozlišujte medzi selektívnym deficitom IgA, t.j. pozostávajúce z nedostatku jednej z podtried (30 % prípadov) a úplného (70 % prípadov). Nedostatok v podtriede IgA2 vedie k výraznejšiemu klinickému obrazu ako nedostatok v podtriede IgA1. Možné sú aj kombinácie deficitu IgA s inými poruchami: s poruchou biosyntézy IgG a s abnormalitami T-lymfocytov. Prevažná väčšina ľudí so selektívnym
Nedostatok IgA je prakticky zdravý. Pre deti do 2 rokov je nedostatok IgA fyziologickým stavom.
Zníženie koncentrácie IgA v sére na<5 мг/дл у детей старше 4 лет; IgG и IgM в норме, количество и соотношение субпопуляций лимфоцитов и их функциональная активность могут быть в норме.
Klinický obraz. Pri nedostatku IgA sa môžu vyvinúť 3 skupiny patologických syndrómov: infekčné, autoimunitné a alergické. Pacienti s deficitom IgA sú náchylní na opakované infekcie horných dýchacích ciest a tráviaceho systému. Najčastejšími a najzávažnejšími sú rôzne autoimunitné ochorenia (reumatoidná artritída, ankylozujúca spondylitída, Sjogrenov syndróm, vaskulitída s poškodením mozgových ciev, autoimunitná tyreoiditída, SLE, glomerulonefritída, hemolytická anémia, diabetes mellitus I. typu, vitiligo a pod.) Výskyt celiakie je 10-krát vyšší ako u detí s normálnym IgA. Najčastejšie zistené alergické prejavy: intolerancia na bielkovinu kravského mlieka, atopická dermatitída (AD), bronchiálna astma.
Liečba. Asymptomatické prípady nevyžadujú žiadnu špeciálnu liečbu; v prítomnosti klinických prejavov infekčných, autoimunitných a alergických ochorení sa liečba uskutočňuje v súlade s normami.
Substitučná liečba darcovskými imunoglobulínmi nie je indikovaná ani pri selektívnom, ani pri úplnom deficite IgA, pretože je vysoká pravdepodobnosť tvorby antiizotypových protilátok proti IgA u príjemcu a rozvoj transfúznych komplikácií nimi spôsobených.
Agamaglobulinémia s nedostatkom B-buniek
X-viazaná agamaglobulinémia (Brutonova choroba) predstavuje 90 % všetkých prípadov agamaglobulinémie. Chlapci, synovia (אּ, ρ) nosičov defektného génu sú chorí btk (Xq21.3-q22), kódujúca proteín tyrozínkinázu špecifickú pre B-lymfocyty Btk (Brutonova tyrozínkináza- Brutonova tyrozínkináza). V dôsledku defektu dochádza k porušeniu intracelulárnych signálnych dráh, rekombinácii ťažkých reťazcov imunoglobulínov,
renováciu pre B-buniek na B-lymfocyty. U 10 % pacientov sa agamaglobulinémia s deficitom B-buniek dedí autozomálne recesívnym spôsobom. V súčasnosti je popísaných 6 genetických defektov, vrátane molekúl pre-B-bunkového receptora, cytoplazmatického B-bunkového adaptérového proteínu (BLNK) a génu Opakovanie bohaté na leucín 8 (LRRC8).
Údaje z laboratórneho výskumu. Neexistujú žiadne periférne B-lymfocyty. Kostná dreň obsahuje pre-B bunky s μ reťazcom v cytoplazme. Počet T lymfocytov a funkčné testy T lymfocytov môžu byť normálne. IgM a IgA v krvi nemožno zistiť; IgG môže byť prítomný, ale v malých množstvách (0,4-1,0 g / l). Neexistujú žiadne protilátky proti antigénom krvných skupín a vakcínovým antigénom (tetanus, difterické toxíny a pod.). Môže sa vyvinúť neutropénia. Histologické vyšetrenie lymfoidného tkaniva: v lymfoidných folikuloch nie sú žiadne germinálne (embryonálne) centrá a plazmatické bunky.
Klinický obraz. Ak rodinná anamnéza nie je známa, diagnóza sa stane zrejmou v priemere vo veku 3,5 roka. Ochorenie je charakterizované hypopláziou lymfatického tkaniva, ťažkými hnisavými infekciami, infekčnými ochoreniami horných (sinusitída, zápal stredného ucha) a dolných (bronchitída, pneumónia) dýchacích ciest; možná gastroenteritída, pyodermia, septická artritída (bakteriálna alebo chlamýdiová), septikémia, meningitída, encefalitída, osteomyelitída. Najčastejšími pôvodcami ochorení dýchacích ciest sú Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus, hnačka črevné baktérie alebo giardia Giardia lamblia. Pacienti s agamaglobulinémiou sú tiež náchylní na infekčné ochorenia spôsobené mykoplazmami a ureaplazmami, ktoré sú príčinou rozvoja chronickej pneumónie, purulentnej artritídy, cystitídy a abscesov podkožného tkaniva. Typické pre vírusy sú neurotropné vírusy ECHO-19 a coxsackie, ktoré spôsobujú ťažkú akútnu aj chronickú encefalitídu a encefalomyelitídu. Entero prejavy vírusové infekcie môže sa vyskytnúť syndróm podobný dermatomyozitíde, ataxia, bolesti hlavy, poruchy správania. U chorých detí, keď sú imunizované živou vakcínou proti detskej obrne, sa spravidla zistí predĺžené vylučovanie vírusu poliomyelitídy cez sliznice a s obnovenou a zvyšujúcou sa virulenciou (t.j.
existuje reálne nebezpečenstvo nákazy zdravých detí poliomyelitídou v dôsledku kontaktu s očkovaným imunodeficientným dieťaťom). Autoimunitné poruchy pri agamaglobulinémii môžu predstavovať reumatoidná artritída, syndróm podobný sklerodermii, skleredém, ulcerózna kolitída, diabetes mellitus I. typu (v dôsledku prevahy Th1 imunitnej odpovede).
Fyzikálne vyšetrenie. Dávajte pozor na zaostávanie vo fyzickom vývoji, na tvar prstov (prsty vo forme paličiek), zmeny tvaru hrudníka, charakteristické pre choroby dolných dýchacích ciest, hypopláziu lymfatické uzliny a mandle.
Liečba.
Substitučná terapia: intravenózne imunoglobulínové prípravky sa podávajú každé 3-4 týždne po celý život. Dávky imunoglobulínov sa vyberajú tak, aby sa vytvorila ich koncentrácia v sére pacienta presahujúca spodnú hranicu vekovej normy.
Diskutujte o možnosti génovej terapie – gen Btk klonovaný, ale jeho nadmerná expresia je spojená s malígnou transformáciou hematopoetického tkaniva.
V prípade pretrvávajúcej neutropénie sa používajú rastové faktory. Keď sa objavia príznaky autoimunitnej patológie, je možné predpísať monoklonálne protilátky (infliximab atď.).
Bežná variabilná imunitná nedostatočnosť
Všeobecná variabilná imunitná nedostatočnosť (CVID) je skupina syndrómov charakterizovaných poruchou syntézy protilátok a bunkovej imunity. Spoľahlivým diagnostickým kritériom pre CVID je významný pokles obsahu imunoglobulínov dvoch alebo troch hlavných izotypov u oboch pohlaví v kombinácii s jedným z nasledujúcich príznakov:
Nástup choroby vo veku nad 2 roky;
Nedostatok izohemaglutinínov a/alebo nízka odpoveď na očkovanie;
Vylúčenie iných príčin agamaglobulinémie.
U niektorých pacientov sú príčinou rozvoja CVID mutácie v génoch kódujúcich molekuly zapojené do procesov dozrievania a prežívania B buniek: BAFF-R (receptor aktivačného faktora B-buniek), Blimp-1 (B-lymfocytmi indukovaný maturačný proteín-1) a ICOS (Indukovateľný kostimulátor). Dochádza k narušeniu schopnosti B-lymfocytov diferencovať sa na plazmatické bunky, vznikajú poruchy tvorby protilátok, je možná dysfunkcia T-lymfocytov a pozoruje sa zvýšený sklon k infekčným ochoreniam. Syndróm sa môže prejaviť v ranom detstve, dospievaní alebo u mladých dospelých.
Údaje z laboratórneho výskumu. Hladiny IgG a IgA (asi u 50 % pacientov) a IgM (až do nedetegovateľných množstiev) sú výrazne znížené. Počet B-lymfocytov v krvi je normálny alebo znížený. Počet T-lymfocytov u väčšiny pacientov je normálny. U ťažkých pacientov sa môže vyvinúť lymfopénia (menej ako 1500x103 buniek v 1 litri krvi). Počet NK buniek je znížený. Produkcia špecifických protilátok v reakcii na imunizáciu je znížená alebo chýba. Proliferácia lymfocytov a tvorba IL-2 pod vplyvom mitogénov a antigénov sú výrazne narušené.
Klinický obraz. Zisťujú sa recidivujúce bakteriálne infekčné ochorenia s lokalizáciou najmä v dýchacích cestách a vedľajších nosových dutinách. V čase diagnózy môžu infekcie dýchacích ciest progredovať do bronchiektázie a difúznych pľúcnych lézií. Možná infekčná lézia tráviaceho systému, prejavujúca sa hnačkou, steatoreou a malabsorpciou (a teda úbytkom hmotnosti). Infekcie spôsobené Giardia lamblia, Pneumocystis carinii alebo vírusy rodiny Herpetoviridae. Pacienti s CVID sú náchylní na rozvoj purulentnej artritídy spôsobenej mykoplazmami a ureaplazmami. Prejavy enterovírusových infekcií môžu byť encefalomyelitída, poliomyelitída a syndrómy podobné dermatomyozitíde, lézie kože a slizníc. Autoimunitné choroby sú ťažké a môžu určiť prognózu CVID. Niekedy sú prvými klinickými prejavmi CVID artritída, ulcerózna kolitída a Crohnova choroba, sklerotizujúca cholangitída, malabsorpcia, SLE, nefritída, myozitída, autoimunitné ochorenie pľúc vo forme lymfoidnej intersticiálnej pneumonitídy, neutropénia,
trombocytopenická purpura, hemolytická anémia, perniciózna anémia, alopécia totalis, retinálna vaskulitída, fotosenzitivita. U pacientov s CVID je významne zvýšený výskyt (v 15 % prípadov) sarkoidóze podobných granulómov a nemalígnej lymfoproliferácie. Liečba.
Antibakteriálna chemoterapia.
Substitučná terapia: intravenózne imunoglobulínové prípravky sa podávajú každé 3-4 týždne po celý život.
V prípade autoimunitných komplikácií - imunosupresívna liečba (glukokortikoidy, azatioprín, cyklosporín A) a prípadne vymenovanie monoklonálnych protilátok (infliximab atď.).
Hyper-IgM syndrómy
Hyper-IgM syndrómy sú pomerne zriedkavé ochorenia charakterizované výrazným znížením alebo úplnou absenciou IgG, IgA a normálnou alebo zvýšenou koncentráciou IgM v sére. Je to spôsobené neschopnosťou B-lymfocytov prepínať triedy imunoglobulínov a hypermutagenézou variabilných domén. Doteraz bolo identifikovaných 6 genetických defektov, ktoré vedú k rozvoju hyper-IgM syndrómu.
. Typ 1 (HIGM 1). X-viazaný deficit CD40 ligandu (70 % prípadov syndrómov hyper-IgM), čo vedie k neschopnosti T buniek účinne interagovať s B lymfocytmi.
. Typ 2 (HIGM 2). Autozomálne recesívne, spojené s defektom v AID-indukovanej aktivácii cytidíndeaminázy (gén Aicda, 12p13)- enzým podieľajúci sa na prepínaní tried imunoglobulínov a hypermutagenéze.
. Typ 3 (HIGM 3). Autozomálne recesívne, spojené s génovou mutáciou molekuly CD40. Zároveň samotné B-bunky nie sú schopné účinne interagovať s T-lymfocytmi. Fenotypové prejavy sú podobné prejavom 1. typu.
. Typ 4 (HIGM 4). Autozomálne recesívne; v niektorých prípadoch dochádza k mutáciám de novo. Súvisí s defektom UNG - uracil-DNA glykozylázy - enzýmu, ktorý sa tiež podieľa
pri prepínaní tried imunoglobulínov, ale po pôsobení AID. V tomto prípade nie je ovplyvnená hypermutagenéza a syndróm prebieha s menšou závažnosťou.
. Typ 5 (HIGM 5). Chyba len pri prepínaní tried, hypermutagenéza nie je ovplyvnená. Kauzálna mutácia ešte nebola identifikovaná, ale zrejme existuje defekt v enzýme pôsobiacom po
AID.
. Typ 6 (HIGM-ED). X-viazaná, spojená s dyshidrotickou ektodermálnou dyspláziou, je spôsobená nedostatkom NEMO (NF-kB modulátor), čo má za následok zhoršenú signalizáciu CD40.
X-viazaný hyper-IgM syndróm identifikované častejšie ako iné. Vyvíja sa s defektom v géne kódujúcom CD40L (CD154, gén sa nachádza na Xq26-q27.2) je ligand pre CD40. Nedostatočná expresia CD40L T-lymfocytmi vedie k nemožnosti zámeny tried imunoglobulínov v B-lymfocytoch z IgM na iné izotypy, ako aj k poruche tvorby pamäťových B-buniek, repertoáru T-buniek a odpovede Th1-buniek namierených proti intracelulárne mikroorganizmy. Chlapci sú chorí
Údaje z laboratórneho výskumu. IgG, IgA, IgE sa nedajú určiť alebo sa detegujú vo veľmi malých množstvách. Hladiny IgM sú normálne (v 50 % prípadov) alebo zvýšené, často výrazne. Počet T a B buniek je normálny; znížená proliferatívna odpoveď T buniek, indukovaná antigénmi. IgM sú polyklonálne, niekedy monoklonálne. Odhaliť autoprotilátky izotypu IgM (antierytrocytové, protidoštičkové, antityroidné, protilátky proti antigénom tkaniva hladkého svalstva). V lymfoidnom tkanive nie sú žiadne zárodočné centrá, ale existujú plazmatické bunky.
Klinický obraz. Prvé prejavy sa vyskytujú v detstve a ranom detstve. Opakované infekcií odlišná lokalizácia (predovšetkým dýchacie cesty), vrátane oportúnnych (spôsobených Pneumocystis carinii). Charakteristické sú aj vírusové lézie (cytomegalovírusy a adenovírusy), Criptococcus neoformans, mykoplazmy a mykobaktérie. Kryptosporidiová infekcia môže spôsobiť akútnu a chronickú hnačku (rozvíja sa u 50 % pacientov) a sklerotizujúcu cholangitídu. Často sa rozvíja anémia, neutropénia, ulcerácia ústnej sliznice, zápal ďasien, ulcerózna
lézie pažeráka, rôznych častí čreva, ulcerózna kolitída. Prezrádzajú predispozíciu k autoimunitné poruchy(séronegatívna artritída, glomerulonefritída atď.) a malígne novotvary (hlavne lymfatické tkanivo, pečeň a žlčové cesty). Môže sa vyvinúť lymfadenopatia, hepato- a splenomegália. Liečba
Pravidelná substitučná liečba intravenóznym imunoglobulínom.
Antibakteriálna chemoterapia. Na prevenciu a liečbu pneumónie spôsobenej Pneumocystis sa používa kotrimoxazol [sulfametoxazol + trimetoprim] a pentamidín.
Aby ste predišli poškodeniu pečene a žlčových ciest, používajte iba prevarenú alebo filtrovanú vodu, vykonajte pravidelné vyšetrenia (ultrazvuk, biopsia pečene, ak je to indikované).
Pri liečbe neutropénie a ulcerácie ústnej dutiny sa používajú glukokortikoidy a prípravky faktora stimulujúceho kolónie granulocytov.
S rozvojom autoimunitných komplikácií je predpísaná imunosupresívna liečba (glukokortikoidy, azatioprín, cyklosporín A), ako aj lieky na báze monoklonálnych protilátok.
Optimálna liečba je transplantácia kostnej drene od HLA-zhodných darcov (miera prežitia 68 %, najlepšie je vykonať do 8. roku života).
KOMBINOVANÉ IMUNODEFICIE S PREVENTÍVNYM PORUCHOM T-LYMFOCYTOV
Ťažká kombinovaná imunitná nedostatočnosť
SCID (SCID - Ťažká kombinovaná imunitná nedostatočnosť)- skupina syndrómov charakterizovaná poklesom hladiny T-lymfocytov alebo ich úplnou absenciou a poruchou adaptačnej imunity. ... Retikulárna dysgenéza, charakterizovaná poruchou dozrievania lymfoidných a myeloidných prekurzorov v skorých štádiách: neutropénia a T - B - NK -.
. X-viazaný SCID, ktorý sa vyvíja v dôsledku génovej mutácie IL-2RG[(CD132, celkom pri- receptorový reťazec pre IL-2, IL-4, IL-7, IL-9, IL-15 a IL-21), Xq13.1-q21.1,אּ ], čo vedie k blokáde receptorov a neschopnosti cieľových buniek reagovať na pôsobenie zodpovedajúcich interleukínov (viac ako 50 % všetkých prípadov SCID); T - B + NK -.
. Deficit tyrozínkinázy Janus3 [gen JAK3 (19p13.1),ρ ]; s génovými defektmi, prenos aktivačného signálu od všeobecn pri-reťazce IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 do bunkového jadra, čo vedie k zhoršenej diferenciácii T a NK buniek; T - B + NK -.
. Nedostatok proteín tyrozín fosfatázy (CD45, gen PTPRC, 1q31-q32); s génovým defektom dochádza k zvýšeniu inhibičnej aktivity Csk kinázy na proteín tyrozín kinázu Src s poruchou fosforylácie ITAM domén TCR a BCR; T - B + NK +.
. Úplný deficit enzýmov RAG1 a RAG2, ktoré aktivujú rekombináciu V (D) J-segmentov imunoglobulínov a TCR [gény RAG1 a RAG2 (11p13),ρ ]; T - B - NK +.
. Omennov syndróm (neúplný nedostatok RAG1 a
RAG2) [gény RAG1 a/alebo RAG2 (11p13-p12),R]. Vďaka
nízka zvyšková aktivita týchto enzýmov stále vytvára množstvo klonov T-lymfocytov, špecifických pre antigény epitelových tkanív kože a tráviaceho traktu, kde sa množia a produkujú veľké množstvá IL-4 a IL-5, čo spôsobuje hypereozinofília a tvorba IgE reziduálnymi B-lymfocytmi (pri absencii iných tried imunoglobulínov). Charakterizovaná erytrodermiou a pachydermou s alopéciou v pokožke hlavy a obočia, vyčerpávajúcou hnačkou, život ohrozujúcim infekčným syndrómom; hepatosplenomegália a hyperplázia lymfatických uzlín.
. SCID so zvýšenou citlivosťou na ionizujúce žiarenie. Defekt jadrového proteínu Artemis [gen DCLRE1C, (10p),R], zahrnuté v komplexe enzýmov potrebných na opravu DNA (podieľa sa na spojení dvojvláknových zlomov), pri génovej mutácii dochádza k porušeniu V (D) J-rekombinácie; T - B - NK +.
. Nedostatok IL-2 [gen IL-2, 4q26-q27].
Mutácie v géne a-reťazca IL-2 receptora (CD25) (10p15-p14);T - B + NK +.
Mutácie v géne a-reťazca receptora IL-7 (CD127) (5p13);T - B + NK +.
Nedostatok TAP (Transportér na prezentáciu antigénu), nevyhnutné pre transport antigénnych peptidov do endoplazmatického retikula génu a-reťazca IL-7 receptora (CD127) (5p13);T - B + NK +.
Mutácie génov reťazca CD3 (CD3γ, CDδ a CDε), vedúce k zníženiu počtu zrelých T-lymfocytov, zhoršeniu ich diferenciácie; T - B + NK +.
Nedostatok proteín tyrozín kinázy ZAP-70 [gén ZAP-70 (2q12), R]. Pri génovej mutácii trpí fosforylácia ITAM domén ζ-reťazca TCR a receptorov NK buniek obsahujúcich ITAM a vzniká selektívny deficit CD8+ T buniek (obsah CD4+ T lymfocytov je normálny, ale funkčný poruchy sú vyjadrené vo forme absencie IL-2 a proliferácie).
Nedostatok adenozíndeaminázy [gen ada (20q12-q13.11 , p)], čo vedie k akumulácii metabolitov v bunkách (deoxyadenozíntrifosfát a S-adenosyl homocysteín), ktoré inhibujú proliferáciu T- a B-lymfocytov (popísané sú varianty s neskorým nástupom ochorenia); T - B - NK -.
Nedostatok purín nukleozid fosforylázy [gen pnp (14q11.2), p], čo vedie k akumulácii deoxyguanozíntrifosfátu v bunkách, čo inhibuje proliferáciu T-lymfocytov (súbežné syndrómy – urikémia a urikúria); T - B + NK -.
Údaje z laboratórneho výskumu. Odhaliť variabilnú, niekedy hlbokú lymfopéniu; lymfocyty nie sú schopné proliferovať ako odpoveď na špecifický antigén; často sa prejavuje zníženie hladiny imunoglobulínov v krvnom sére. Na röntgenovom snímku hrudníka nie je žiadny tieň týmusu.
Klinický obraz. Zvyčajne sa klinická diagnóza vyjasní v prvých 6 mesiacoch života, keď matky IgG protilátky vymiznú. V klinickom obraze vystupujú do popredia ťažký infekčný syndróm, hypoplázia lymfoidného tkaniva a oneskorenie vývoja. Infekčný syndróm je charakterizovaný orálnou kandidózou, chronickou hnačkou, zápalom pľúc, horúčkou,
sepsa bakteriálnej etiológie, vírusové infekcie. Pôvodcovia infekcií patria do rôznych taxonomických skupín: baktérie, vírusy, huby, oportúnne mikroorganizmy (Pneumocystis carinii). Pneumónia je často spôsobená P. carinii, hnačka - rotavírusy, Campylobacter, Giardia lamblia.Často sa prejavuje vírusová hepatitída. Charakteristický je rozvoj regionálnej alebo generalizovanej BCGitídy po očkovaní.
Liečba zabezpečuje vymenovanie podpornej terapie vrátane parenterálnej výživy, zavedenie intravenózneho imunoglobulínu, vymenovanie antibiotík, antifungálnych a antivírusových liekov. Jednou z hlavných metód liečby na dosiahnutie uzdravenia je transplantácia kostnej drene, bez ktorej deti so SCID zvyčajne zomierajú v 1. roku života. Sú opísané ojedinelé prípady, keď dieťa v obzvlášť sanitovaných podmienkach žilo až 2-3 roky. Je dôležité rozpoznať SCID u novorodencov čo najskôr, pretože napríklad imunizácia živými vakcínami je pre nich smrteľná. Ihneď po stanovení diagnózy je potrebné takéto deti umiestniť do gnotobiologických podmienok (sterilný box). V prípade pridania infekčných ochorení sa vykonáva intenzívna antibakteriálna, antivírusová a antifungálna terapia, substitučná liečba intravenóznym imunoglobulínom. Na prevenciu pneumocystovej pneumónie je predpísaný kotrimoxazol. Ak sa rozvinie BCGitída, je potrebné vykonávať dlhodobú intenzívnu antituberkulóznu liečbu. Na transfúziu zložiek krvi by sa mali používať iba ožiarené a filtrované lieky. Existuje riziko potransfúznej reakcie štepu proti hostiteľovi v dôsledku transplacentárneho prenosu materských lymfocytov.
Syndróm nahých lymfocytov
Toto je názov patológie, keď molekuly MHC-I alebo MHC-II nie sú exprimované v tele. Pri absencii expresie molekúl MHC-I je obsah CD8+ T-lymfocytov znížený a aktivita NK buniek chýba; v neprítomnosti MHC-II je znížená hladina CD4 + T-lymfocytov. Bolo charakterizovaných niekoľko genetických defektov. Tieto defekty však nie sú lokalizované v génoch MHC, ale v niekoľkých rôznych faktoroch zodpovedných za ich reguláciu
výraz. Klinický obraz syndróm „nahých“ lymfocytov a liečbe sú podobné ako pri iných SCID.
DiGeorgov syndróm
S DiGeorgeovým syndrómom alebo syndrómom defektu tretieho a štvrtého faryngálneho vrecka [vypustenie v 22q11, vrátane génu TBX1 (22q11.2), odhaliť hypopláziu alebo apláziu týmusu, hypopláziu prištítneho telieska, srdcové chyby, deficit T-lymfocytov, premenlivý počet B-lymfocytov.
Údaje z laboratórneho výskumu. Významný pokles počtu CD3+, CD4+ a CD8+ T buniek a prudký pokles ich proliferačnej aktivity vyvolanej mitogénmi a antigénmi. Počet B a NK buniek je normálny. Koncentrácia sérových imunoglobulínov je vo väčšine prípadov v medziach normy, sú možné rôzne varianty dysgamaglobulinémie.
Klinický obraz. Imunodeficientnú zložku predstavuje hypoplázia alebo aplázia týmusu a recidivujúca, ťažká infekčné choroby. Odhalia aj hypoparatyreózu (hypokalciémiu a následkom toho tetániu, viditeľnú 1. – 2. deň po narodení); defekty obehového systému (pravé otočenie oblúka aorty, stenóza pravej komory, defekty medzikomorových a interatriálnych sept, Fallotova tetrada, atrézia alebo hypoplázia pľúcnej tepny); dutina podnebia; anomálie tvárového skeletu (zväčšená vzdialenosť párových orgánov, zmenšené čeľuste, najmä dolné, nízko nasadené ušnice, krátka nosová ryha). Vyjadrené anomálie v štruktúre hrtana, hltana, priedušnice, vnútorného ucha, pažeráka; zhoršený vývoj obličiek, centrálneho nervového systému a iné malformácie (polydaktýlia, absencia nechtov, atrézia konečníka, análne fistuly). Charakteristické je oneskorenie reči a psychomotorického vývinu. Zaznamenávajú predispozíciu k autoimunitné poruchy(cytopénia, autoimunitná tyroiditída) a malígne novotvary.
Liečba.... Antibakteriálna a antivírusová terapia. ... Substitučná liečba intravenóznymi imunoglobulínovými prípravkami. ... Chirurgická liečba na korekciu malformácií. ... Pri autoimunitných komplikáciách - imunosupresívna liečba. ... V prítomnosti endokrinopatií korekcia zodpovedajúcich porúch. ... Transplantácia kostnej drene je neúčinná
tivna. ... Transplantácia epiteliálneho tkaniva týmusu je opodstatnená. Korekcia funkcie prištítnych teliesok.
X-viazaný lymfoproliferatívny syndróm
NS- spojený lymfoproliferatívny syndróm je charakterizovaný poruchou imunitnej odpovede na vírus Epstein-Barrovej [v dôsledku génových defektov SH2D1A(SAP) v Xq25,אּ], čo vedie k nekontrolovanej proliferácii B-lymfocytov transformovaných vírusom Epstein-Barrovej a infekcii nových cieľových buniek vírusom.
Klinický obraz. Sú opísané 4 najčastejšie fenotypy: ťažká infekčná mononukleóza, malígne lymfoproliferatívne stavy (lymfómy, leukémie, hlavne B-bunky), anémia alebo pancytopénia (vrátane hemofagocytárneho syndrómu vyvolaného vírusom), dysgamaglobulinémia. Infekcia vírusom Epstein-Barr je spúšťacím (spúšťacím) mechanizmom vzniku najťažších, rýchlo progresívnych a smrteľných ochorení: fulminantná infekčná mononukleóza (smrteľná v 58% prípadov), hemofagocytárny syndróm (bez liečby v 100% prípadov prípady, smrteľné). V 10% prípadov sa fenotyp prejavuje pred infekciou vírusom Epstein-Barrovej (spravidla sa vyvíja dysgamaglobulinémia a lymfómy). Najčastejšie sa zisťujú rôzne typy hypogamaglobulinémie. Imunodeficiencia vedie k rozvoju bakteriálnych, plesňových a vírusových infekčné choroby. Je možné predpokladať ochorenie u chlapcov s charakteristickou rodinnou anamnézou a séro- alebo PCR pozitívnym testom na vírus Epstein-Barrovej. Na diagnostiku sa odporúča použiť kombináciu genetickej analýzy SH2D1A a hodnotenie úrovne expresie SAP.
Liečba
Pre účely prevencie sa odporúča užívať antivírusové lieky - acyklovir, valaciklovir (ich skoré podanie potláča replikáciu vírusu Epstein-Barrovej v orofaryngu) a intravenózny imunoglobulín (s vysokým titrom protilátok proti vírusu Epstein-Barrovej ).
Pri hypogamaglobulinémii sa intravenózny imunoglobulín používa mesačne v kombinácii s antibiotickou terapiou.
Na liečbu fulminantnej infekčnej mononukleózy sú predpísané vysoké dávky acykloviru a metylprednizolónu, vysokodávková terapia intravenóznym imunoglobulínom s vysokým titrom protilátok proti vírusu Epstein-Barrovej a IFNa.
S rozvojom hemofagocytárneho syndrómu sa vysoké dávky dexametazónu kombinujú s vepezidom ♠ (etopozid).
Pri liečbe malígnych ochorení sa používajú štandardné terapeutické protokoly.
Radikálnou liečebnou metódou je transplantácia kostnej drene od HLA zodpovedajúcich darcov.
Autoimunitný lymfoproliferatívny syndróm
Autoimunitný lymfoproliferatívny syndróm - skupina ochorení charakterizovaná benígnou lymfoproliferáciou, hyperimunoglobulinémiou, autoimunitnými poruchami, zvýšeným obsahom CD3 + CD4 - CD8 - T-lymfocytov (dvojito negatívnych) v periférnej krvi a poruchou apoptózy [defekt génu Fas(CD95) - TNFRSF6 (10q24.1), gén kaspáza-10, Fas ligand - FasL (1q23)].
Údaje z laboratórneho výskumu. Obsah CD3 + CD4 - CD8 - T-lymfocytov v periférnej krvi alebo lymfatických tkanivách je viac ako 1%. Hladiny IgG, IgA a IgM môžu byť normálne, zvýšené alebo dokonca znížené. Vekom je hypergamaglobulinémia nahradená nízkou koncentráciou sérových imunoglobulínov až po agamaglobulinémiu. Odhalenie autoprotilátok proti erytrocytom, krvným doštičkám, neutrofilom, hladkým svalom a faktoru VIII; antinukleárne a antifosfolipidové autoprotilátky, ako aj reumatoidný faktor atď. Charakteristická je lymfocytóza.
Klinický obraz. Všetci pacienti majú zväčšenú pečeň, lymfatické uzliny (v prvých 5 rokoch života) a slezinu. Lymfoproliferáciu nesprevádza horúčka a nočné potenie. Debut autoimunitné reakcie sa nemusí zhodovať s lymfoproliferáciou a vyskytuje sa neskôr. S vekom sa závažnosť autoimunitných reakcií zvyšuje. Častejšie sa vyvíjajú autoimunitné reakcie proti krvinkám (hemolytická anémia, trombocytopénia, neutropénia), menej často sú postihnuté iné orgány. Zvyšuje sa riziko vzniku malígnych novotvarov (T- a B-lymfómy, Burkittov lymfóm, atypický lymfóm, lymfogranulomatóza atď.).
Liečba.... Chemoterapeutické látky (cyklofosfamid, azatioprín, metotrexát, chlorambucil). ... Glukokortikoidy. ... Splenektómia s ťažkým hypersplenizmom a hemocytopéniou. ... V závažných prípadoch je možná transplantácia kostnej drene.
Syndróm hyperimunoglobulinémie E
Hyper-IgE syndróm je charakterizovaný výrazným zvýšením hladiny sérového IgE, opakovanými abscesmi kože a podkožia stafylokokovej etiológie, pneumóniou s tvorbou pneumokély, anomáliami v štruktúre tvárového skeletu, AD. Molekulárno-genetická povaha syndrómu hyper-IgE ešte nebola stanovená. V niektorých prípadoch bola odhalená autozomálne dominantná dedičnosť, v iných - autozomálne recesívna dedičnosť. Predpokladá sa, že defekty ovplyvňujú signálne molekuly cytokínových receptorov (pri autozomálne dominantnej forme tohto syndrómu mutácie v Stat3) a prípadne spojené s narušením fungovania subpopulácie Th17 buniek. Ďalší gén zodpovedný za vznik hyper-IgE syndrómu sa nachádza na 4. chromozóme (4q).
Údaje z laboratórneho výskumu. Zisťujú sa rôzne imunologické poruchy: zvýšenie hladiny IgE v sére, porušenie chemotaxie neutrofilov, porucha tvorby protilátok; zníženie odpovede HRT na kandidín, difterický a tetanový toxoid; oslabenie proliferačnej aktivity T buniek v reakcii na Candida a tetanový toxoid pri zachovaní odpovede na mitogény. Eozinofília v periférnej krvi a tekutine kožných abscesov. Počet T a B buniek je normálny.
Klinický obraz. Stredný ekzém v ranom veku. Charakteristické črty tváre (široký nos, široký tupý nos, asymetria tvárového skeletu, vyčnievajúce čelo, hlboko posadené oči, vysoké podnebie). Odhalia anomálie vo vývoji kostry, skoliózu, zvýšenú pohyblivosť kĺbov, sklon k zlomeninám kostí po drobných úrazoch, porušenie náhrady zubov. Vyskytujú sa abscesy kože, podkožia a lymfatických uzlín. Pneumónia sa vyvíja vo vyššom veku (najčastejšie patogény S. aureus a H. in-
chrípka), v 77% prípadov sa vytvorí pneumokéla, infekcia v dôsledku P. aeruginosa a A. fumigatus. Pneumónia môže prebiehať bez horúčky. Chronická kandidóza slizníc a nechtov sa vyvíja v 83% prípadov.
Liečba.... Dlhodobá (za účelom prevencie - doživotná) antibakteriálna a antimykotická terapia. ... Na liečbu dermatitídy sa používajú lokálne látky, v závažných prípadoch nízke dávky cyklosporínu A. Transplantácia kostnej drene je neúčinná.
SYNDRÓMY CHROMOZÓMOVÉHO ZLYHANIA
Pre syndrómy s chromozomálnou nestabilitou: ataxiateleangiektázia[defekt v géne DNA topoizomerázy bankomat (11q22), p] a Nijmegen syndróm[Defekt nibrínového génu NBS1(8q21)] - charakterizované zvýšenou frekvenciou malígnych nádorov, spontánnou chromozomálnou nestabilitou a chromozomálnymi rozpadmi. Oba proteíny sa podieľajú na oprave zlomov dvojvláknovej DNA a na regulácii bunkového cyklu. Normálne sa zlomy dvojvláknovej DNA vyskytujú počas V (D) J rekombinácie imunoglobulínových a TCR génov, pri prepínaní tried imunoglobulínov, počas prechodu a počas meiózy. Podobné procesy sa vyskytujú počas dozrievania neurónov v mozgu. Poruchy opravy DNA pri ataxii-telangiektázii a syndróme Nijmegen spôsobujú také klinické prejavy, ako sú poruchy syntézy imunoglobulínov, funkcie pohlavných orgánov a nervového systému.
Ataxia-telangiektázia
Tento syndróm (frekvencia 1: 300 tisíc novorodencov) s veľmi heterogénnym fenotypom opísal francúzsky lekár D. Louis-Bar. Príznaky ataxie možno u dieťaťa zistiť už vo veku 2-4 mesiacov. Ataxia je spôsobená progresívnou degeneráciou Purkyňových buniek v mozočku. Teleangiektázie na koži nosa, ušníc a spojovky sa objavujú o niečo neskôr, vo veku 3-6 rokov. Na koži sa často objavujú škvrny po káve s mliekom. Charakteristická je hypoplázia týmusu, lymfatických uzlín, sleziny, mandlí. Imunodeficiencia sa prejavuje znížením (často nerovnováhou) tvorby IgA, IgE, IgG2, IgG4. 80% pacientov sa vyvinie
existuje zodpovedajúca infekčná klinická symptomatológia. Znížený počet a funkčná aktivita T buniek (hlavne CD4+ T buniek). Celkový počet T lymfocytov je u väčšiny pacientov normálny. Frekvencia novotvarov (hlavne lymfómov a karcinómov) je nezvyčajne vysoká (200-krát vyššia ako v bežnej populácii), čo často vedie k úmrtiu do 10-12 rokov. Liečba symptomatická.
Nijmegen syndróm
Nijmegen syndróm (pod názvom mesta v Holandsku, kde bola choroba prvýkrát popísaná) sa prejavuje mikrocefáliou, špecifickými poruchami tvárového skeletu (šikmé čelo, vystupujúca stredná časť tváre, dlhý nos, hypoplázia dolnej čeľuste, Mongoloidný očný rez, epikantus, veľké uši), zaostávajúci fyzický vývoj , prítomnosť škvŕn "káva s mliekom" na koži; klinodaktýlia a syndaktýlia, dysgenéza vaječníkov atď. Väčšina detí trpí recidivujúcimi a chronickými bakteriálnymi infekčné choroby dýchacie cesty, orgány ORL a močový systém. V 50 % prípadov sa vyvinú malígne novotvary, najmä B-bunkové lymfómy. Odhaliť rôzne formy dysgamaglobulinémie, pokles CD4 + T buniek.
Liečba.... Symptomatická liečba neurologických porúch. ... Substitučná liečba intravenóznym imunoglobulínom. ... Podľa indikácií sa používa antibakteriálna, antivírusová, antifungálna terapia. ... Pri liečbe malígnych novotvarov sa berie do úvahy zvýšená citlivosť na ožarovanie a chemoterapiu.
Wiskott-Aldrichov syndróm
Wiskott-Aldrichov syndróm [defekt génu WASP (Xp11.23p11.22),אּ; aj ρ a Ʀ] Gen WASP(od Wiskott-Aldrichov syndróm) exprimovaný v lymfocytoch, tkanive sleziny a tymocytoch. Mutácie v tomto géne sú spojené s abnormálnou expresiou v neutrofiloch a T-lymfocytoch (CD4 a CD8) molekuly CD43 (ligand pre ICAM-1, má antiadhéznu funkciu).
Údaje z laboratórneho výskumu. Trombocytopénia (menej ako 10% normy) je spôsobená zvýšenou deštrukciou buniek.
Krvné doštičky sú menšie ako u zdravých ľudí. Hladina IgM v sére je znížená s normálnou hladinou IgG a zvýšením IgA a IgE. Znížené titre izohemaglutinínov, narušená tvorba protilátok proti polysacharidovým antigénom pneumokokov, streptokokov, Escherichia coli, Salmonella, ako aj antivírusových protilátok. V ranom veku je spravidla počet lymfocytov normálny, po 6 rokoch sa zistí lymfopénia (menej ako 1x109 / l), pokles CD3 + a CD4 + T buniek s normálnou hladinou B a NK bunky. Je možná eozinofília a rozvoj posthemoragickej anémie. Proliferatívna odpoveď T buniek na mitogény a antigény klesá, HRT je oslabená. V slezine nie sú zistené normálne štruktúry zárodočných centier a zón T-buniek.
Klinický obraz. Ochorenie je charakterizované triádou symptómov: trombocytopénia, ekzém a opakujúce sa infekčné choroby. Hemoragický syndróm sa prejavuje skoro, už v novorodeneckom období (petechiálna vyrážka, kefalohematómy, krvácanie z pupočnej rany, črevné krvácanie). Ekzém sa prejavuje už od útleho veku u 80 % pacientov. Známky imunodeficiencie sa zvyšujú s vekom: bakteriálne infekčné ochorenia orgánov ORL, dýchacieho systému, tráviacich orgánov, kože; bežná alebo generalizovaná herpetická infekcia (Herpes simplex a Varicella zoster), cytomegalovírus, ako aj plesňové (kandidóza slizníc), menej často oportúnne infekčné ochorenia. Autoimunitné ochorenia (hemolytická anémia, neutropénia, artritída, kožná vaskulitída, ulcerózna kolitída, cerebrálna vaskulitída, glomerulonefritída, autoimunitná trombocytopénia) sú diagnostikované u 70 % pacientov. U pacientov nad 5 rokov je výskyt zhubné novotvary(hlavne nádory lymfatického tkaniva).
Liečba.... Alogénna transplantácia kostnej drene alebo kmeňových buniek (úspešnosť operácie dosahuje 90 % pri použití štepu od histokompatibilného darcu a 50 % pri haploidentickej transplantácii). ... Substitučná liečba intravenóznym imunoglobulínom. ... Preventívne podávanie antibakteriálnych, antimykotických a antivírusových liekov. ... Na zníženie hemoragického syndrómu sa vykonáva splenektómia. ... Pri autoimunitných komplikáciách je predpísaná imunosupresívna liečba.
VADY FAGOCYTÓZY
Chronická granulomatózna choroba
Chronické granulomatózne ochorenie je charakterizované poruchou funkčnej aktivity fagocytov (tvorba aktívnych foriem kyslíkových radikálov, intracelulárne zabíjanie a fragmentácia fagocytovaných patogénov), perzistujúce bakteriálne a plesňové infekčné ochorenia a rozvoj granulomatózneho zápalu. Chronické granulomatózne ochorenie sa vyvíja u osôb s rôznymi genetickými defektmi [v 65 % prípadov - X-viazaný variant ochorenia: gén gp91-phox (Xp21.1),אּ; v 35% prípadov - autozomálne recesívne: gen f47-phox (7q11.23),ρ; gén p67-phox (1q25),ρ; gén p22-phox (16q24), p], čo vedie k poruchám v systéme NADP-oxidázy. So smrťou krátkodobých (niekoľko hodín) neutrofilov "tečú" nezabité baktérie do ohniska zápalu. Makrofágy sú bunky s dlhou životnosťou a ich prekurzory (monocyty) migrujú do ohniska vo zvýšenom počte (čo vedie k vzniku granulómy), fagocytózne mikroorganizmy, ale nie sú schopné ich zabíjať.
Údaje z laboratórneho výskumu. Sérové imunoglobulíny a subpopulácie lymfocytov sú normálne. Tvorba peroxidových radikálov neutrofilmi, hodnotená v testoch (chemiluminiscencia závislá od luminolu alebo redukcia tetrazóliovej modrej), je výrazne znížená alebo chýba. Na pozadí infekčných ochorení je charakteristická leukocytóza, neutrofília, zvýšená ESR, anémia a hypergamaglobulinémia.
Klinický obraz. Ochorenie sa vo väčšine prípadov prejavuje v prvom roku života. infekčný syndróm(infekcie intra- a extracelulárnymi patogénmi) a tvorbu granulómov. Najtypickejšie: poškodenie pľúc (recidivujúce zápaly pľúc, poškodenie hílových lymfatických uzlín, pľúcne abscesy, hnisavá pleuristika), tráviaceho traktu, kožné abscesy a lymfadenitída. Najbežnejšími patogénmi sú kataláza-pozitívne mikroorganizmy: S. aureus, Aspergillus spp.,črevné gramnegatívne baktérie (E. coli, Salmonella spp., Serratia marcescens), menej často - Burkholderia cepacia a Nocardia farcinica. Charakteristický je vývoj pečeňových a subfrenických abscesov, osteomyelitídy, pararektálnych abscesov, sepsy.
Najťažšou, život ohrozujúcou infekčnou komplikáciou je aspergilóza, ktorá sa môže vyskytnúť vo forme difúzneho poškodenia pľúc a iných orgánov (tukového tkaniva, mozgu, kostí, kĺbov, endokardu). U pacientov s chronickým granulomatóznym ochorením po BCG vakcinácii sa často vyvinie infekcia súvisiaca s vakcínou zahŕňajúca regionálne lymfatické uzliny. Mykobakteriálne lézie u pacientov s chronickým granulomatóznym ochorením môžu mať pľúcnu aj extrapulmonárnu lokalizáciu a môžu mať protrahovaný priebeh. Pre pacientov s chronickým granulomatóznym ochorením je charakteristické zaostávanie vo fyzickom vývoji.
Liečba.... Antimikrobiálna terapia: preventívne neustále používanie co-trimoxazolu a antifungálnych liekov (itrakonazol atď.); v prípade infekčných komplikácií sa parenterálne vykonáva kombinovaná antibiotická terapia (2-3 baktericídne antibiotiká, ktoré prenikajú intracelulárne) v kombinácii s antimykotickou liečbou. S rozvojom aspergilózy je indikované dlhodobé užívanie amfotericínu B alebo kaspofungínu. Pri mykobakteriálnej infekcii sa používa kombinácia dlhodobej špecifickej terapie antituberkulóznymi liekmi so širokospektrálnymi antibiotikami. ... Chirurgická liečba je často sprevádzaná hnisaním pooperačnej rany a tvorbou nových ložísk. Možno punkcia drenáž abscesu pod kontrolou ultrazvuku. ... Na liečbu ťažkých infekčných komplikácií s neúčinnosťou antibiotickej terapie je možné použiť granulocytovú masu, vysoké dávky IFNy a G-CSF. ... Transplantácia kostnej drene alebo transplantácia buniek pupočníkovej krvi od kompatibilného súrodenca môže byť úspešná v ranom veku, keď je riziko úmrtia na infekčné komplikácie a ochorenie štep proti hostiteľovi minimálne.
Poruchy adhézie leukocytov
Doteraz boli opísané 3 defekty adhézie leukocytov. Všetci majú
autozomálne recesívna dedičnosť, charakterizovaná rekurentnými a chronickými bakteriálnymi a hubovými infekčnými ochoreniami. Typ I je charakterizovaný absenciou alebo zníženou expresiou CD11/CD18 na leukocytoch, poruchou chemotaxie neutrofilov, leukocytózou (viac ako 25x109), neskorou stratou pupočnej šnúry a rozvojom omfalitídy, zlým hojením rán a absenciou tvorby hnisu v mieste prieniku patogénu do tela.
Liečba.... Antibiotická terapia: infekčné epizódy a profylaktické. ... V závažných prípadoch je predpísaná transplantácia kostnej drene od darcu kompatibilného s HLA.
CHYBY SYSTÉMU KIT
Choroby s nedostatkom komponentov komplementu
Prejavy génových defektov jednotlivých komponentov komplementového systému sú uvedené v tabuľke. 11-2.
Dedičná JSC. Choroby spôsobené nedostatkom komponentov komplementu sú zriedkavo detekované, pretože ich prejav vyžaduje homozygotný stav pre autozomálne alely. Existuje jedna výnimka súvisiaca s C1inh (inhibítor C1 esterázy): génová mutácia C1inh,čo vedie k deficitu inhibítora, v heterozygotnom stave sa prejavuje fenotypom známym ako dedičná AO (podrobnejšie pozri kapitolu 13, Angioedém).
Choroby imunitných komplexov. Nedostatok C1-C4 sa prejavuje rozvojom ochorení imunitných komplexov – systémovej vaskulitídy a poškodenia obličiek, čo sa súhrnne nazýva syndróm systémového lupus erythematosus (SLE).
Pyogénne infekcie. Nedostatok C3 (tiež faktorov H a I) je spojený so zvýšenou náchylnosťou na pyogénne infekcie. Nedostatok zložiek zapojených do alternatívnej dráhy aktivácie komplementu, ako aj nedostatok zložiek C5-C8, sú spojené so zvýšenou náchylnosťou na infekciu spôsobenú Neisseria spp. Nedostatok C9 je zvyčajne klinicky asymptomatický.
Tabuľka 11-2. Klinické prejavy defektov jednotlivých komponentov komplementového systému
Nedostatok lektínu viažuceho manózu
Nedostatok lektínu viažuceho manózu (tiež známy ako lektín viažuci manózu – MSL) je spôsobený génovým defektom MBL(rôzne bodové mutácie a delécie v gen MBL zistené u 17 % ľudí kaukazskej rasy). Pri génových defektoch je narušená aktivácia proteáz, ktoré rozkladajú zložky komplementu C2 a C4, a aktivácia komplementového systému pozdĺž lektínovej dráhy. Klinicky táto patológia sa prejavuje infekčným syndrómom.
Údaje z laboratórneho výskumu. Analýza subpopulácií lymfocytov, leukocytov, ako aj izotypov imunoglobulínov nevykazuje významné odchýlky, adekvátne klinickým symptómom. V krvnom sére nie je MSL.
Liečba. Toto ochorenie nie je klasickou poruchou imunity. Preto je imunokorekcia imunotropnými látkami kontraindikovaná. Rekombinantná MSL sa môže použiť ako farmakologický prostriedok na etiopatogenetickú substitučnú liečbu u pacientov s týmto dedičným defektom. Tento liek v súčasnosti prechádza klinickými skúškami.
Odporúčame tiež
 Matka dievčatka Mauglí bola vyhlásená za šialenú: aká bude ďalšia Irina diagnóza bude tromfom pre opatrovnícke orgány
Matka dievčatka Mauglí bola vyhlásená za šialenú: aká bude ďalšia Irina diagnóza bude tromfom pre opatrovnícke orgány
 Kolika u novorodenca: príznaky, dôvody, ako uľaviť dieťaťu od bolesti, keď prejdú
Kolika u novorodenca: príznaky, dôvody, ako uľaviť dieťaťu od bolesti, keď prejdú
 Ako organizovať nápady pre malé podniky
Ako organizovať nápady pre malé podniky
 DIY dizajn fotoalbumu: neštandardné nápady
DIY dizajn fotoalbumu: neštandardné nápady
 Výber zdroja financovania podnikateľského projektu alebo rozvoja existujúceho malého podniku Vymenujte interné a externé zdroje financovania podnikania
Výber zdroja financovania podnikateľského projektu alebo rozvoja existujúceho malého podniku Vymenujte interné a externé zdroje financovania podnikania
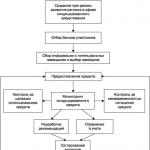 Hlavné zdroje financovania podnikania
Hlavné zdroje financovania podnikania